

Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene
- PMID: 18313022
- PMCID: PMC2427209
- DOI: 10.1016/j.ajhg.2008.01.010
Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene
Abstract
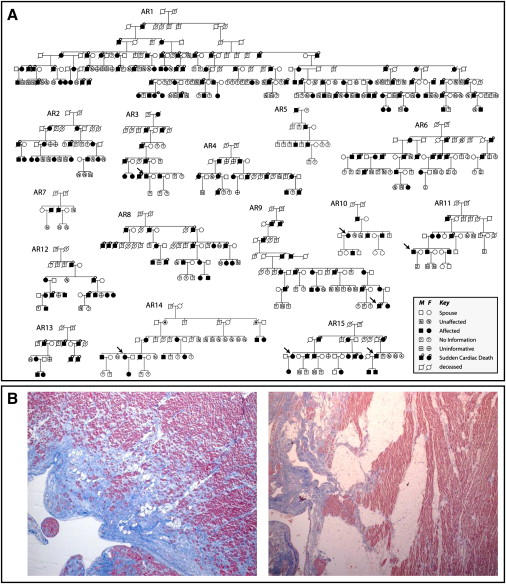
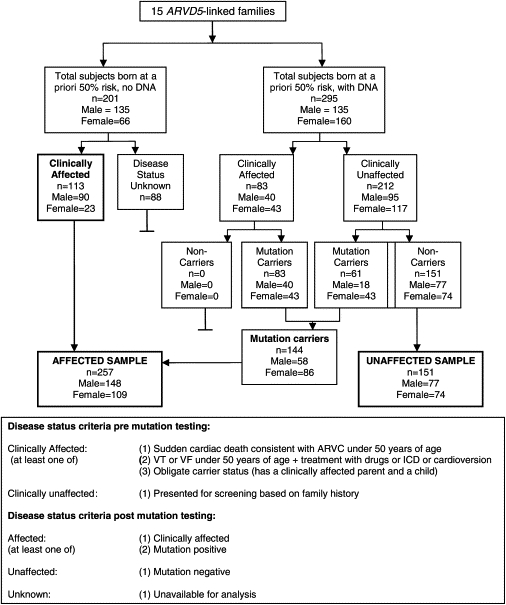
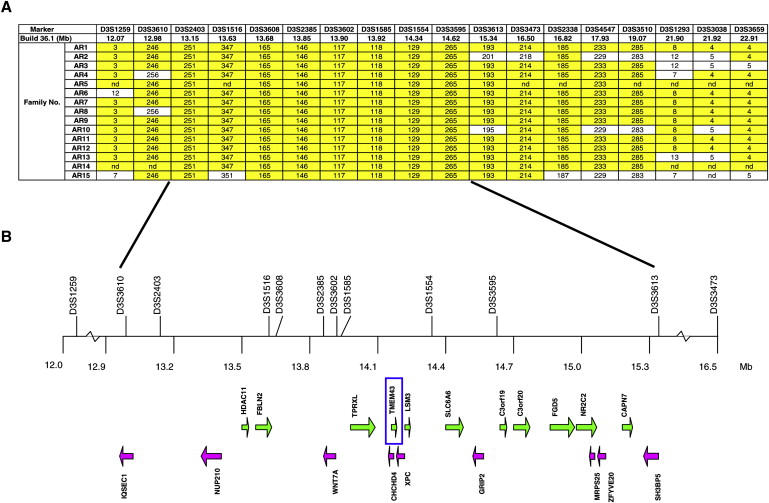
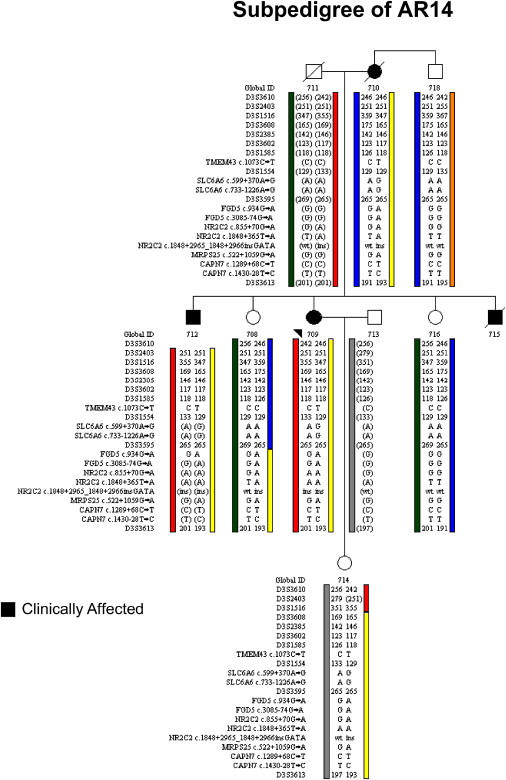
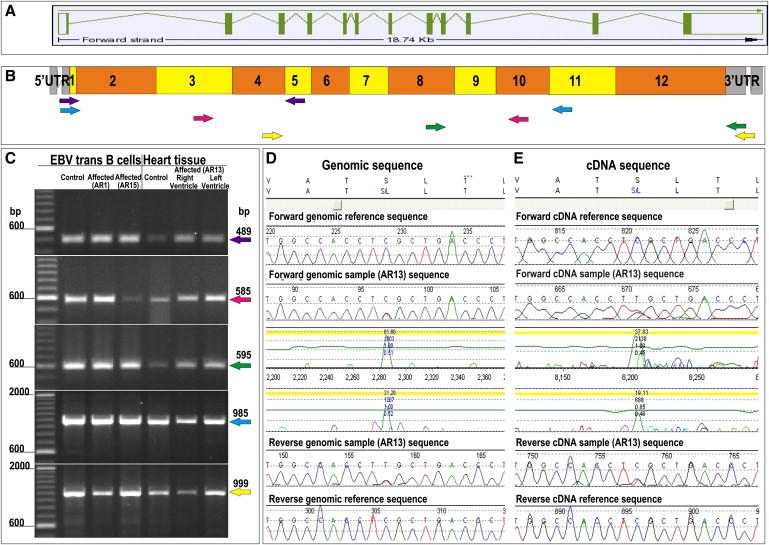
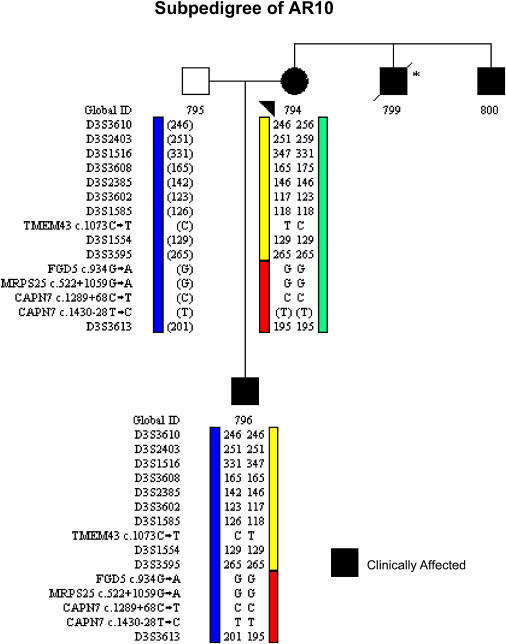
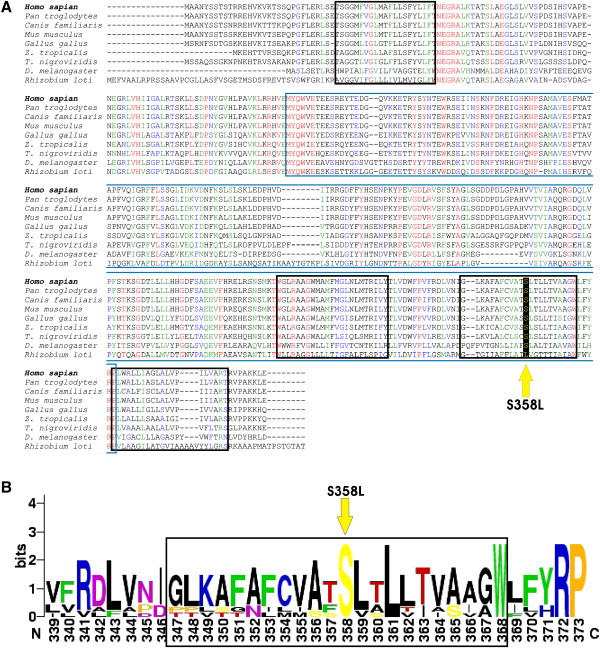
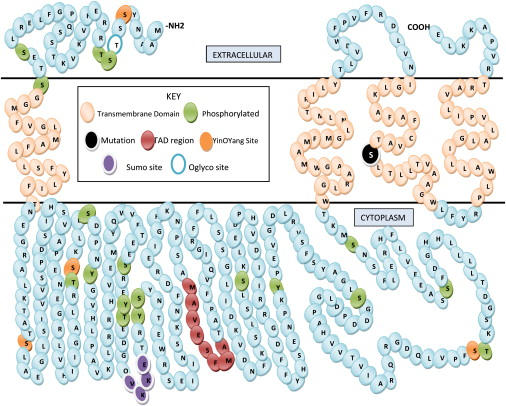
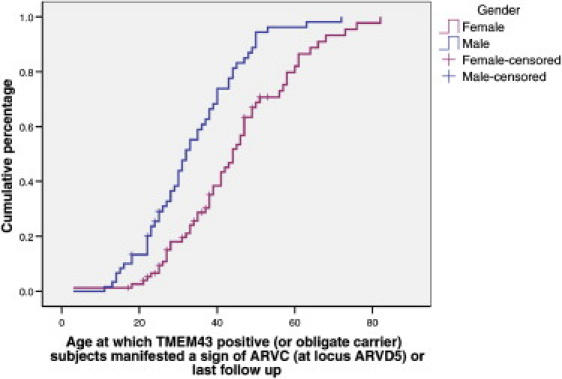
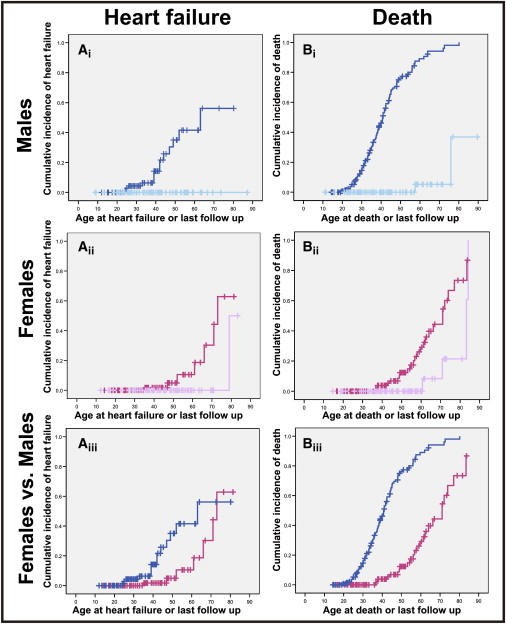
Autosomal-dominant arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) causes sudden cardiac death and is characterized by clinical and genetic heterogeneity. Fifteen unrelated ARVC families with a disease-associated haplotype on chromosome 3p (ARVD5) were ascertained from a genetically isolated population. Identification of key recombination events reduced the disease region to a 2.36 Mb interval containing 20 annotated genes. Bidirectional resequencing showed one rare variant in transmembrane protein 43 (TMEM43 1073C-->T, S358L), was carried on all recombinant ARVD5 ancestral haplotypes from affected subjects and not found in population controls. The mutation occurs in a highly conserved transmembrane domain of TMEM43 and is predicted to be deleterious. Clinical outcomes in 257 affected and 151 unaffected subjects were compared, and penetrance was determined. We concluded that ARVC at locus ARVD5 is a lethal, fully penetrant, sex-influenced morbid disorder. Median life expectancy was 41 years in affected males compared to 71 years in affected females (relative risk 6.8, 95% CI 1.3-10.9). Heart failure was a late manifestation in survivors. Although little is known about the function of the TMEM43 gene, it contains a response element for PPAR gamma (an adipogenic transcription factor), which may explain the fibrofatty replacement of the myocardium, a characteristic pathological finding in ARVC.
Figures
References
-
- Fontaine G., Fontaliran F., Frank R. Arrhythmogenic right ventricular cardiomyopathies: Clinical forms and main differential diagnoses. Circulation. 1998;97:1532–1535. - PubMed
-
- Protonotarios N., Tsatsopoulou A., Anastasakis A., Sevdalis E., McKoy G., Stratos K., Gatzoulis K., Tentolouris K., Spiliopoulou C., Panagiotakos D. Genotype-phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J. Am. Coll. Cardiol. 2001;38:1477–1484. - PubMed
-
- Pilichou K., Nava A., Basso C., Beffagna G., Bauce B., Lorenzon A., Frigo G., Vettori A., Valente M., Towbin J. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricularcardiomyopathy. Circulation. 2006;113:1171–1179. - PubMed
-
- Sen-Chowdhry S., Syrris P., McKenna W. Genetics of right ventricular cardiomyopathy. J. Cardiovasc. Electrophysiol. 2005;16:927–935. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
