

Review
doi: 10.1172/JCI34275.
Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice
Affiliations
- PMID: 18317565
- PMCID: PMC2254980
- DOI: 10.1172/JCI34275
Item in Clipboard
Review
Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice
J Clin Invest.
2008 Mar.
Abstract
Nonalcoholic fatty liver disease (NAFLD) is associated with obesity, insulin resistance, and type 2 diabetes. NAFLD represents a large spectrum of diseases ranging from (i) fatty liver (hepatic steatosis); (ii) steatosis with inflammation and necrosis; and (iii) cirrhosis. Although the molecular mechanism leading to the development of hepatic steatosis in the pathogenesis of NAFLD is complex, recent animal models have shown that modulating important enzymes in fatty acid synthesis in liver may be key for the treatment of NAFLD. This review discusses recent advances in the field.
Figures
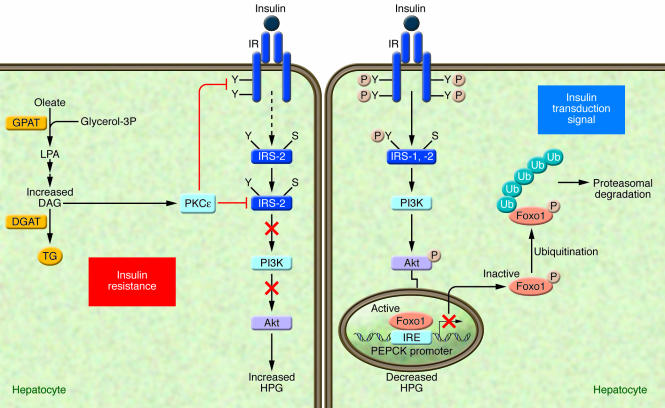
Under conditions in which an adequate transduction signal is present (right panel), insulin binding to the insulin receptor results in the phosphorylation of tyrosine residues (Y) on insulin receptor substrates (IRS-1 and -2), which leads to the activation of PI3K and the subsequent phosphorylation of Akt, which are involved in mediating the metabolic effects of insulin. The transcription factor Foxo1, plays a key role in the regulation of HGP, through the transcriptional control of gluconeogenic enzymes, such as phosphoenolpyruvate carboxykinase (PEPCK). Insulin-mediated Akt phosphorylation of Foxo1 leads to its nuclear exclusion, ubiquitination, and subsequent proteasomal degradation, leading to the decreased PEPCK transcription. In turn, gluconeogenic rates and blood glucose concentrations decrease (14). Under conditions of insulin resistance (HF/HC-diet) (left panel), excess lipid metabolites such as DAG can cause insulin resistance by activating PKCε. The activated PKCε binds to the insulin receptor and inhibits its tyrosine kinase activity. The activation of PKCε may also interfere with the ability of insulin to phosphorylate IRS-2 on tyrosine residues (91). IRE, insulin-responsive element; S, serine; Ub, ubiquitination.
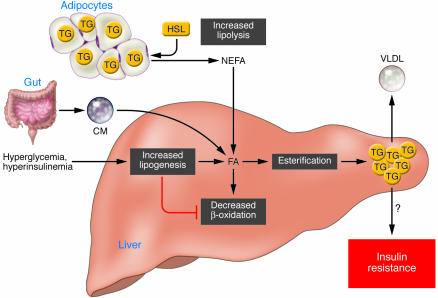
Different sources of fatty acids contribute to the development of fatty liver. Under conditions of insulin resistance, insulin does not adequately inhibit HSL, and lipolysis in white adipose tissue is not suppressed. Therefore peripheral fats stored in adipose tissue flow to the liver by way of plasma NEFAs. Dietary fatty acids are also taken up by the liver through the uptake of intestinally derived chylomicron (CM). In addition, the combination of elevated plasma glucose (hyperglycemia) and insulin concentrations (hyperinsulinemia) promotes de novo fatty acid synthesis (lipogenesis) and impairs β-oxidation, thereby contributing to the development of hepatic steatosis. After the esterification step (conversion of FAs into TGs) TG can then be stored as lipid droplets within hepatocytes or secreted into the blood as VLDL. Although the hepatic accumulation of lipids is widely believed to result in insulin resistance, it remains uncertain whether a causal relationship exists. Several recent studies have even showed a clear dissociation between hepatic steatosis and insulin resistance (7, 87). FA, fatty acid.
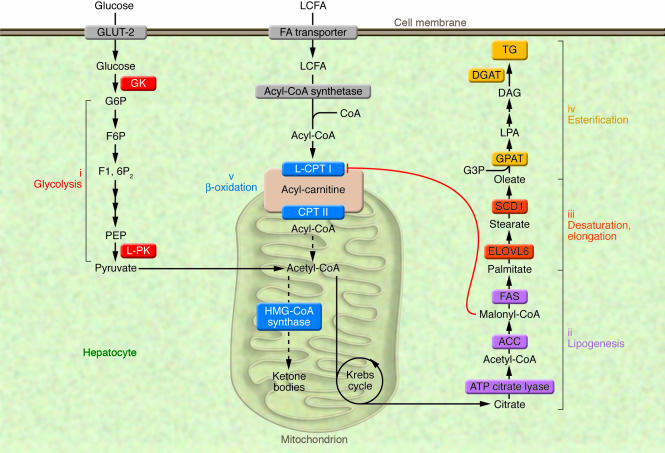
The synthesis of TGs in liver is nutritionally regulated. The ingestion of a LF/HC diet causes a marked induction of enzymes involved in key metabolic pathways, including (i) glucokinase (GK) and L-PK for glycolysis; (ii) ATP citrate lyase, ACC, and FAS for lipogenesis; (iii) ELOVL6 and SCD1 for fatty acid elongation and desaturation steps; and finally (iv) GPAT and DGAT for TG synthesis. Under these nutritional conditions, elevation in malonyl-CoA concentrations, the product of the lipogenic enzyme ACC, inhibits L–CPT I, the rate-limiting enzyme of β-oxidation (v), which regulates the transfer of long-chain acyl-CoAs from the cytosol into the mitochondria, thus resulting in a shift from an oxidative (production of ketone bodies) to an esterification pathway (TG synthesis). F6P, fructose 6-phosphate; F1, 6P2, fructose 1,6 diphosphate; G3P, glycerol 3-phosphate; G6P, glucose 6-phosphatase; PEP, phosphoenol pyruvate; LCFA, long-chain fatty acids; CPT II, carnitine palmitoyltransferase II.
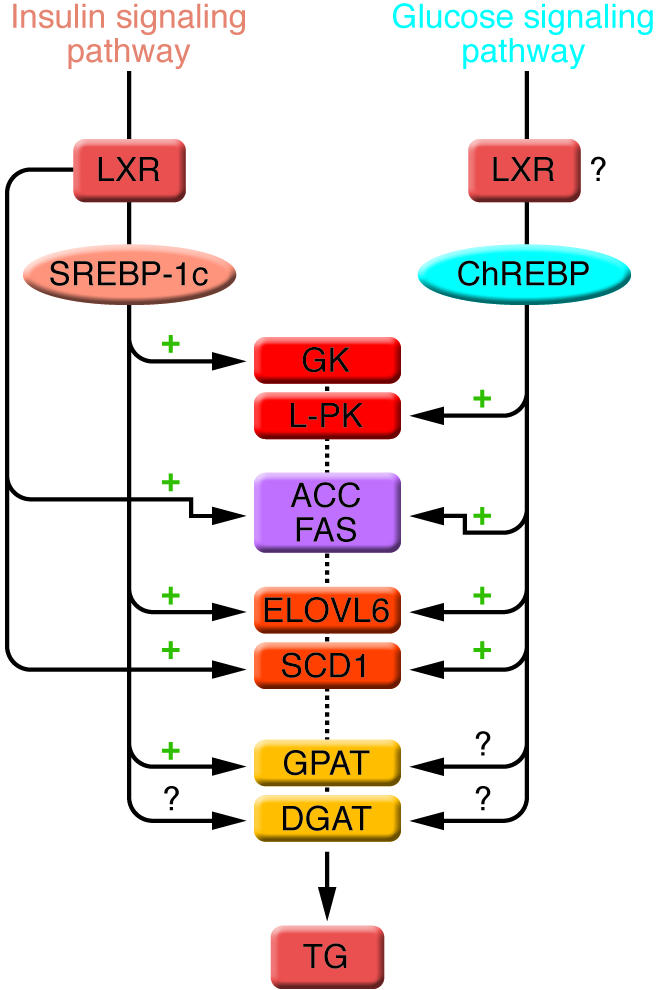
The conversion of glucose into TGs is nutritionally regulated, and both insulin and glucose signaling pathways are activated in response to dietary carbohydrates to synergistically induce gene expression. The transcription factor SREBP-1c mediates the effect of insulin on GK, ACC, FAS, ELOVL6, SCD1, and GPAT. LXRs are also important regulators of TG synthesis, through the direct transcriptional activation of ACC, FAS, and SCD1 but also indirectly via the insulin-mediated transcriptional activation of SREBP1-c. ChREBP, which is induced by glucose, is required for the induction of L-PK, which is exclusively dependent on glucose. The induction of ACC, FAS, ELOVL6, and SCD1 genes is under the synergistic action of ChREBP and SREBP-1c in response to glucose and insulin, respectively. The direct effect of ChREBP on GPAT expression is not clearly established. ChREBP is also a direct transcriptional target of LXRs, and glucose was recently shown to bind and activate LXRs, thereby indicating that LXRs are part of the glucose signaling pathway. However, in our point of view, the relevance of a potential regulation by LXRs of glucose-sensitive genes needs to be demonstrated in a physiological context. The transcriptional regulation of DGAT in liver is, to our knowledge, still largely unknown.
References
-
- Ahmed M.H., Byrne C.D. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD). Drug Discov. Today. 2007;12:740–747. - PubMed
-
- Abdelmalek M.F., Diehl A.M. Nonalcoholic fatty liver disease as a complication of insulin resistance. Med. Clin. North Am. 2007;91:1125–1149. - PubMed
-
- Charlton M. Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin. Gastroenterol. Hepatol. 2004;2:1048–1058. - PubMed
-
- Adams L.A., Lindor K.D. Nonalcoholic fatty liver disease. Ann. Epidemiol. 2007;17:863–869. - PubMed
-
- Yamaguchi K., et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
