

Targeted mutation of mouse skeletal muscle sodium channel produces myotonia and potassium-sensitive weakness
- PMID: 18317596
- PMCID: PMC2260907
- DOI: 10.1172/JCI32638
Targeted mutation of mouse skeletal muscle sodium channel produces myotonia and potassium-sensitive weakness
Abstract
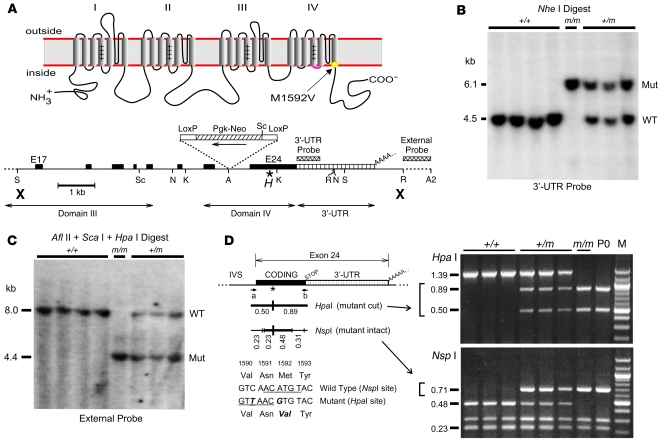
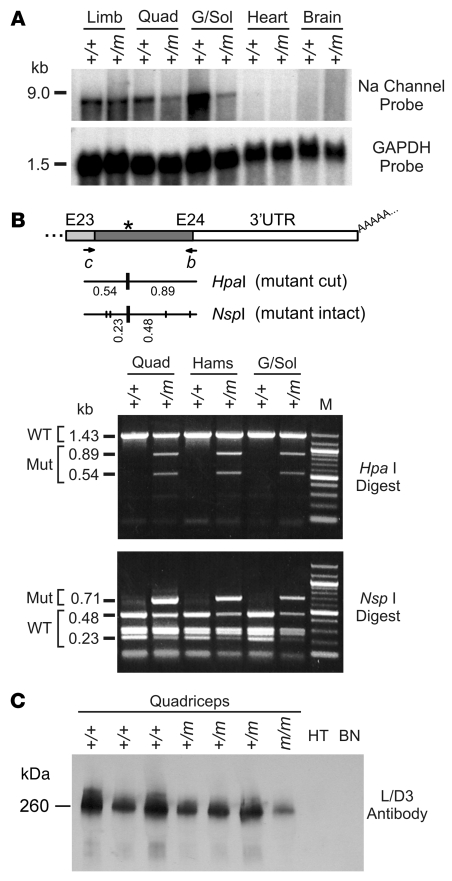
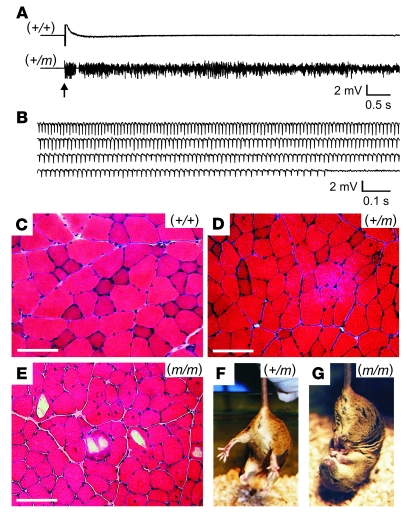
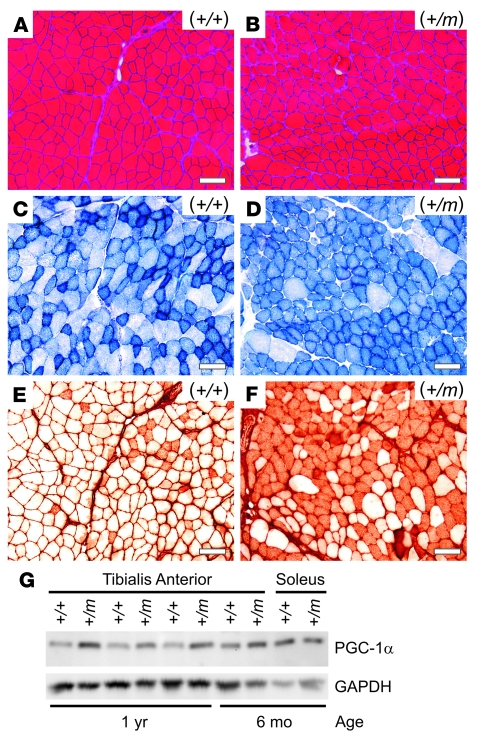
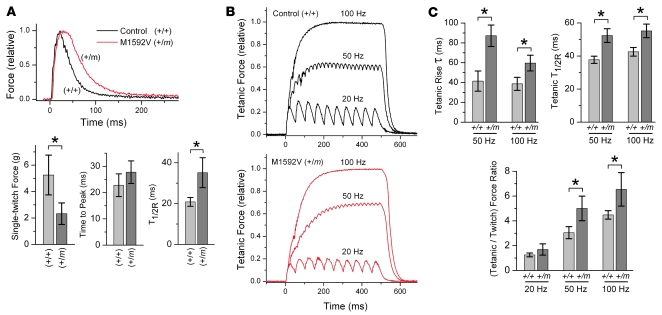
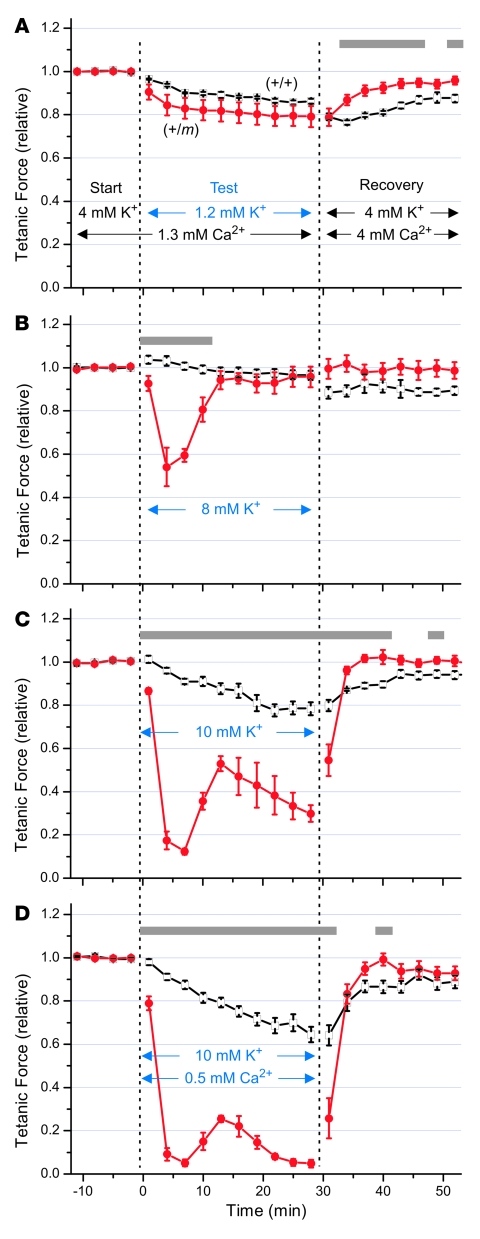
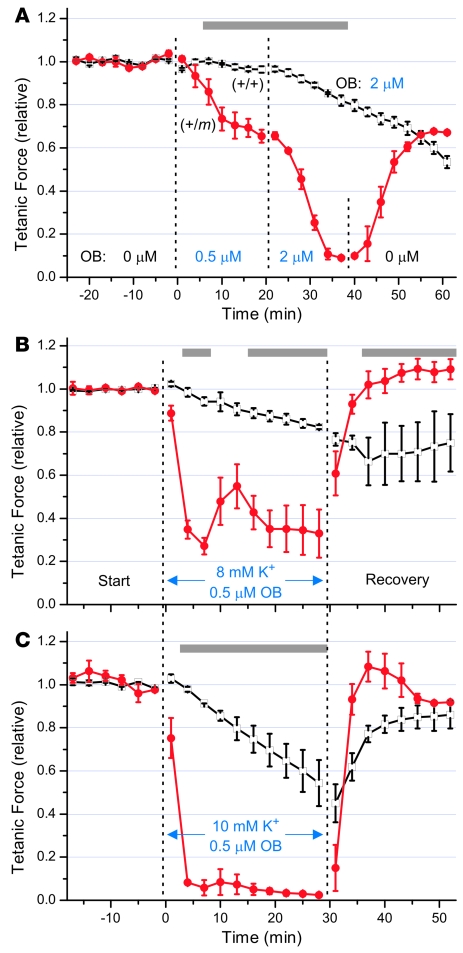
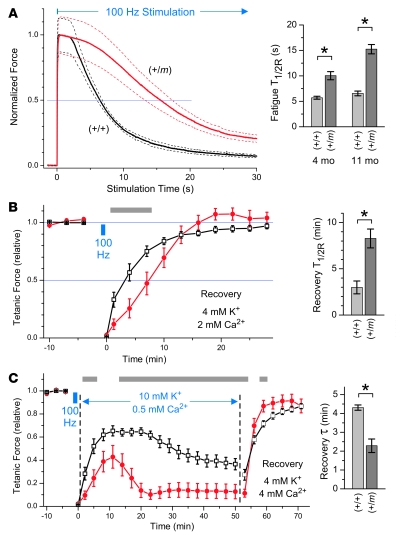
Hyperkalemic periodic paralysis (HyperKPP) produces myotonia and attacks of muscle weakness triggered by rest after exercise or by K+ ingestion. We introduced a missense substitution corresponding to a human familial HyperKPP mutation (Met1592Val) into the mouse gene encoding the skeletal muscle voltage-gated Na+ channel NaV1.4. Mice heterozygous for this mutation exhibited prominent myotonia at rest and muscle fiber-type switching to a more oxidative phenotype compared with controls. Isolated mutant extensor digitorum longus muscles were abnormally sensitive to the Na+/K+ pump inhibitor ouabain and exhibited age-dependent changes, including delayed relaxation and altered generation of tetanic force. Moreover, rapid and sustained weakness of isolated mutant muscles was induced when the extracellular K+ concentration was increased from 4 mM to 10 mM, a level observed in the muscle interstitium of humans during exercise. Mutant muscle recovered from stimulation-induced fatigue more slowly than did control muscle, and the extent of recovery was decreased in the presence of high extracellular K+ levels. These findings demonstrate that expression of the Met1592ValNa+ channel in mouse muscle is sufficient to produce important features of HyperKPP, including myotonia, K+-sensitive paralysis, and susceptibility to delayed weakness during recovery from fatigue.
Figures
References
-
- Lehmann-Horn, F., Rudel, R., and Jurkat-Rott, K. 2004. Nondystrophic myotonias and periodic paralyses.Myology . A.G. Engel and C. Franzini-Armstrong, editors. 3rd edition. McGraw-Hill. New York, New York, USA. 1257–1300.
-
- Gamstorp I., Hauge M., Helweglarsen H.F., Mjones H., Sagild U. Adynamia episodica hereditaria: a disease clinically resembling familial periodic paralysis but characterized by increasing serum potassium during the paralytic attacks. Am. J. Med. 1957;23:385–390. doi: 10.1016/0002-9343(57)90318-2. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
