

Birth-death prior on phylogeny and speed dating
- PMID: 18318893
- PMCID: PMC2270800
- DOI: 10.1186/1471-2148-8-77
Birth-death prior on phylogeny and speed dating
Abstract
Background: In recent years there has been a trend of leaving the strict molecular clock in order to infer dating of speciations and other evolutionary events. Explicit modeling of substitution rates and divergence times makes formulation of informative prior distributions for branch lengths possible. Models with birth-death priors on tree branching and auto-correlated or iid substitution rates among lineages have been proposed, enabling simultaneous inference of substitution rates and divergence times. This problem has, however, mainly been analysed in the Markov chain Monte Carlo (MCMC) framework, an approach requiring computation times of hours or days when applied to large phylogenies.
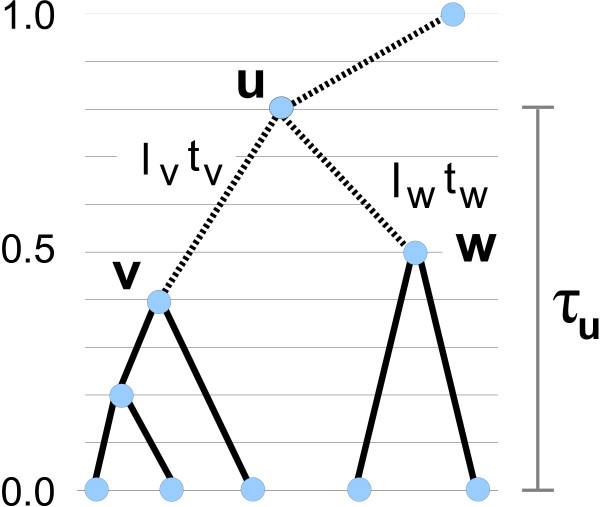
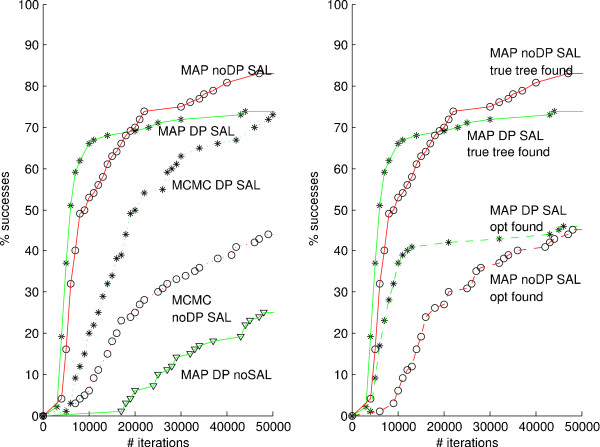
Results: We demonstrate that a hill-climbing maximum a posteriori (MAP) adaptation of the MCMC scheme results in considerable gain in computational efficiency. We demonstrate also that a novel dynamic programming (DP) algorithm for branch length factorization, useful both in the hill-climbing and in the MCMC setting, further reduces computation time. For the problem of inferring rates and times parameters on a fixed tree, we perform simulations, comparisons between hill-climbing and MCMC on a plant rbcL gene dataset, and dating analysis on an animal mtDNA dataset, showing that our methodology enables efficient, highly accurate analysis of very large trees. Datasets requiring a computation time of several days with MCMC can with our MAP algorithm be accurately analysed in less than a minute. From the results of our example analyses, we conclude that our methodology generally avoids getting trapped early in local optima. For the cases where this nevertheless can be a problem, for instance when we in addition to the parameters also infer the tree topology, we show that the problem can be evaded by using a simulated-annealing like (SAL) method in which we favour tree swaps early in the inference while biasing our focus towards rate and time parameter changes later on.
Conclusion: Our contribution leaves the field open for fast and accurate dating analysis of nucleotide sequence data. Modeling branch substitutions rates and divergence times separately allows us to include birth-death priors on the times without the assumption of a molecular clock. The methodology is easily adapted to take data from fossil records into account and it can be used together with a broad range of rate and substitution models.
Figures




References
-
- Stamatakis A, Ludwig T, Meier H. Proceedings of the 2004 ACM Symposium on Applied Computing. ACM Press, New York; 2004. A fast program for Maximum Likelihood-based inference of large phylogenetic trees.
-
- Yang Z, Rannala B. Bayesian phylogenetic inference using DNA sequences: A Markov chain Monte Carlo method. Mol Biol Evol. 1997;14(7):717–724. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
