

Fibrodysplasia ossificans progressiva
- PMID: 18328989
- PMCID: PMC2424023
- DOI: 10.1016/j.berh.2007.11.007
Fibrodysplasia ossificans progressiva
Abstract
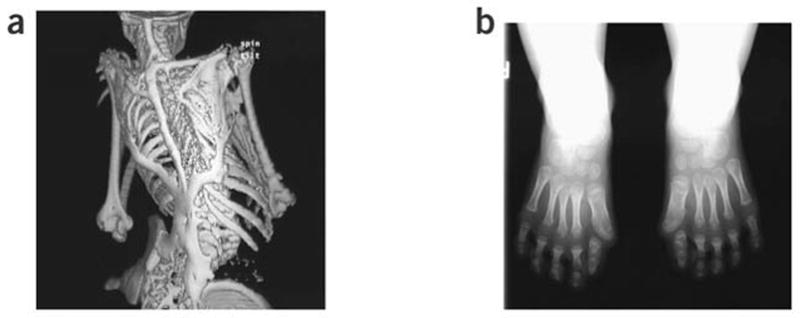
Fibrodysplasia ossificans progressiva (FOP), a rare and disabling genetic condition of congenital skeletal malformations and progressive heterotopic ossification (HO), is the most catastrophic disorder of HO in humans. Episodic disease flare-ups are precipitated by soft tissue injury, and immobility is cumulative. Recently, a recurrent mutation in activin receptor IA/activin-like kinase 2 (ACVR1/ALK2), a bone morphogenetic protein (BMP) type I receptor, was reported in all sporadic and familial cases of classic FOP, making this one of the most highly specific disease-causing mutations in the human genome. The discovery of the FOP gene establishes a critical milestone in understanding FOP, reveals a highly conserved target for drug development in the transforming growth factor (TGF)-beta/BMP signalling pathway, and compels therapeutic approaches for the development of small molecule signal transduction inhibitors for ACVR1/ALK2. Present management involves early diagnosis, assiduous avoidance of iatrogenic harm, and symptomatic amelioration of painful flare-ups. Effective therapies for FOP, and possibly for other common conditions of HO, may potentially be based on future interventions that block ACVR1/ALK2 signalling.
Figures
References
-
- Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history 34 patients. J Bone Joint Surg Br. 1982;64:76–83. - PubMed
-
- Smith R. Fibrodysplasia (myositis) ossificans progressiva: clinical lessons from a rare disease. Clin Orthop Rel Res. 1988;346:7–14. - PubMed
-
- Kaplan FS, Shore EM, Connor JM. Fibrodysplasia ossificans progressiva (FOP) In: Royce PM, Steinmann B, editors. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2. New York: Wiley-Liss, John Wiley & Sons, Inc.; 2002. pp. 827–840.
-
- Kaplan FS, Glaser DL, Hebela N, Shore EM. Heterotopic ossification. J Am Acad Orthop Surg. 2004;12:116–125. - PubMed
-
- Kaplan FS, Glaser DL, Shore EM, et al. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:183–188.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
