

Thrombin
- PMID: 18329094
- PMCID: PMC2491495
- DOI: 10.1016/j.mam.2008.01.001
Thrombin
Abstract
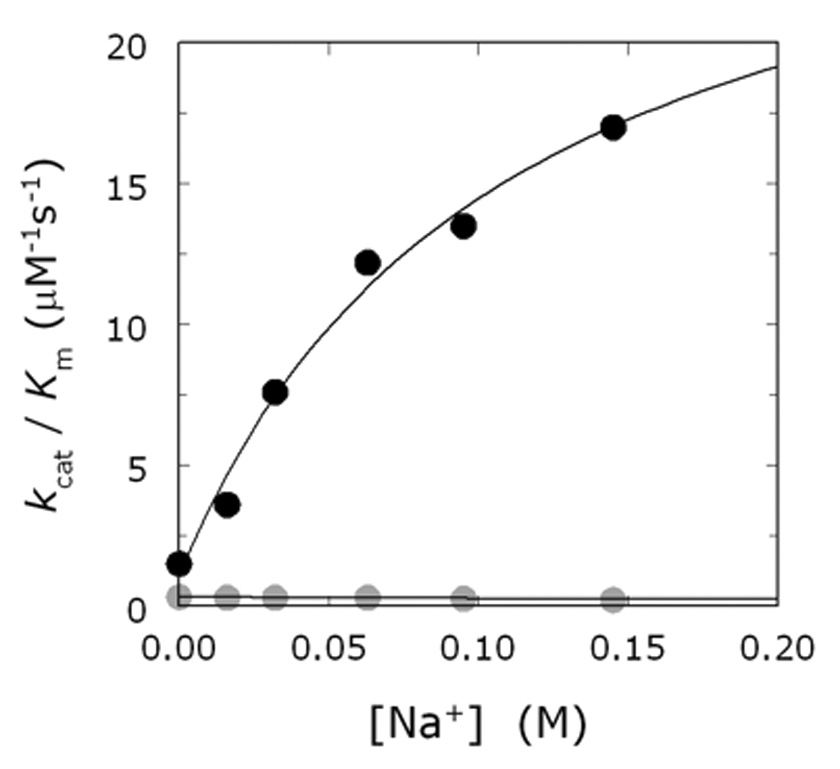
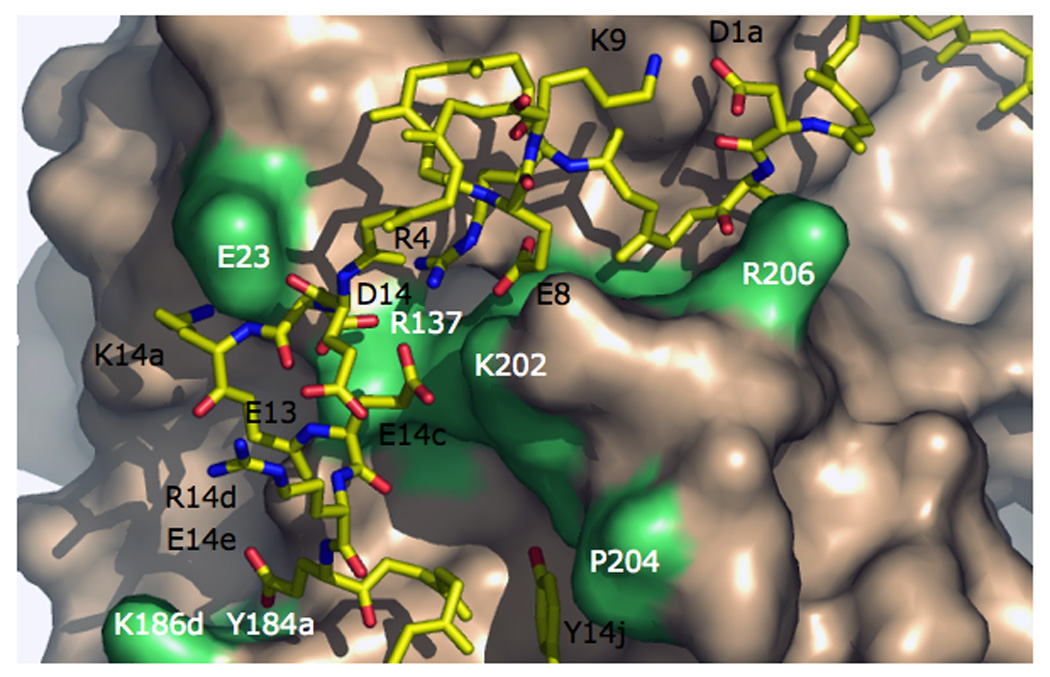
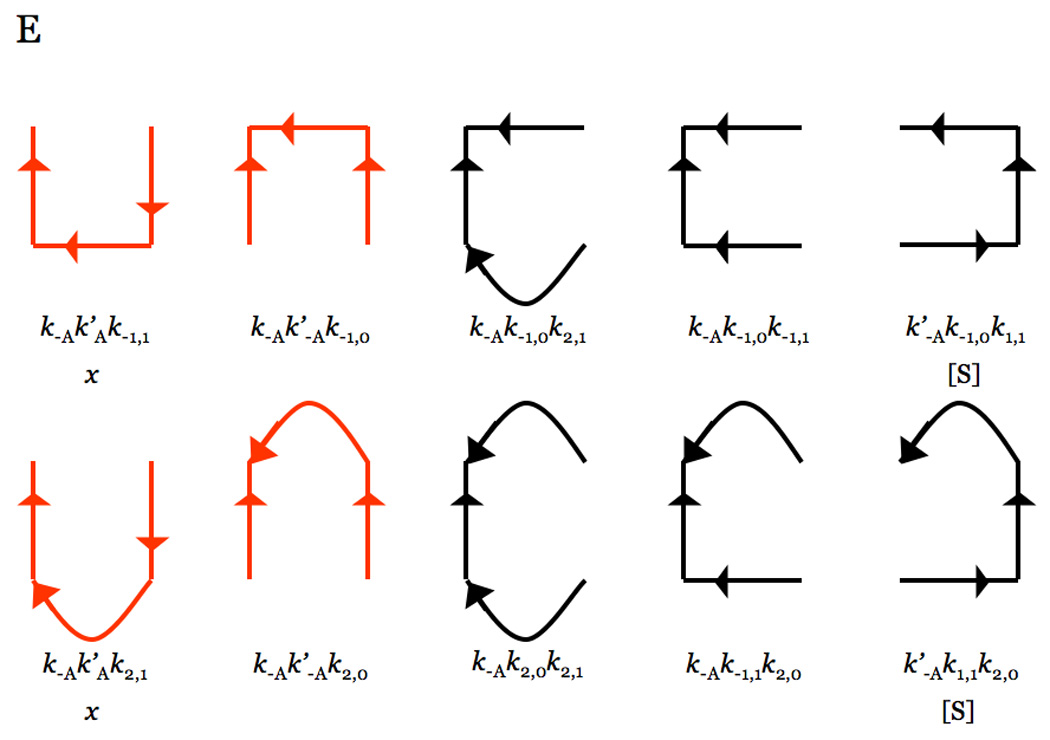
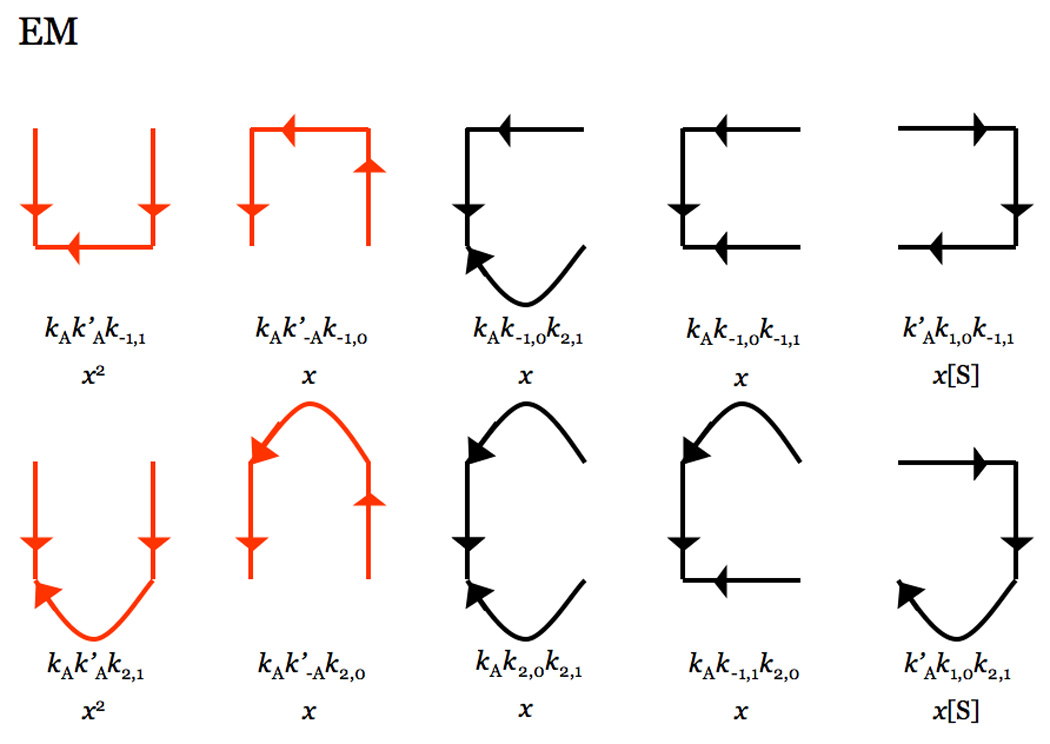
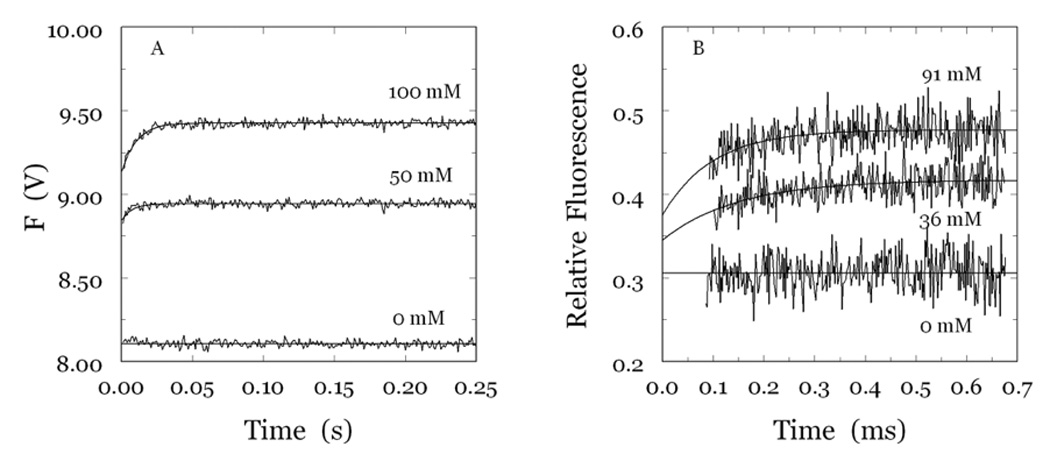
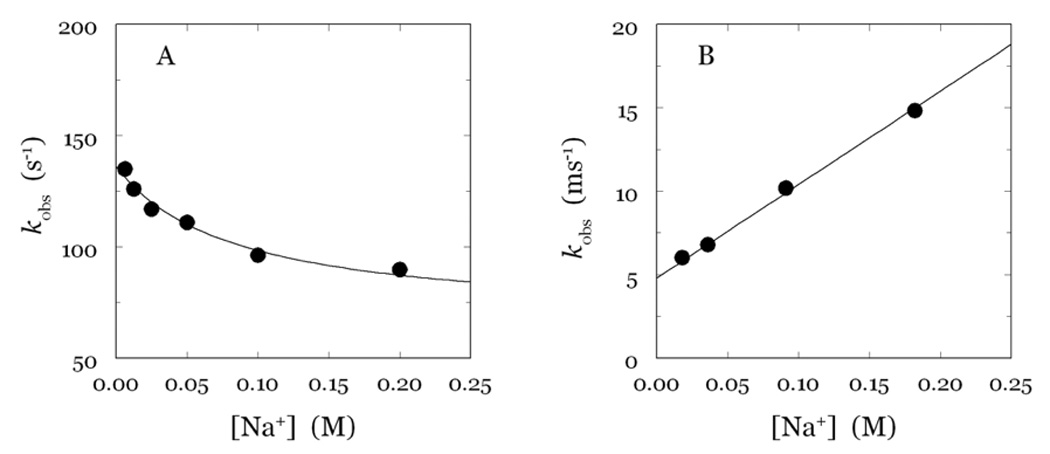
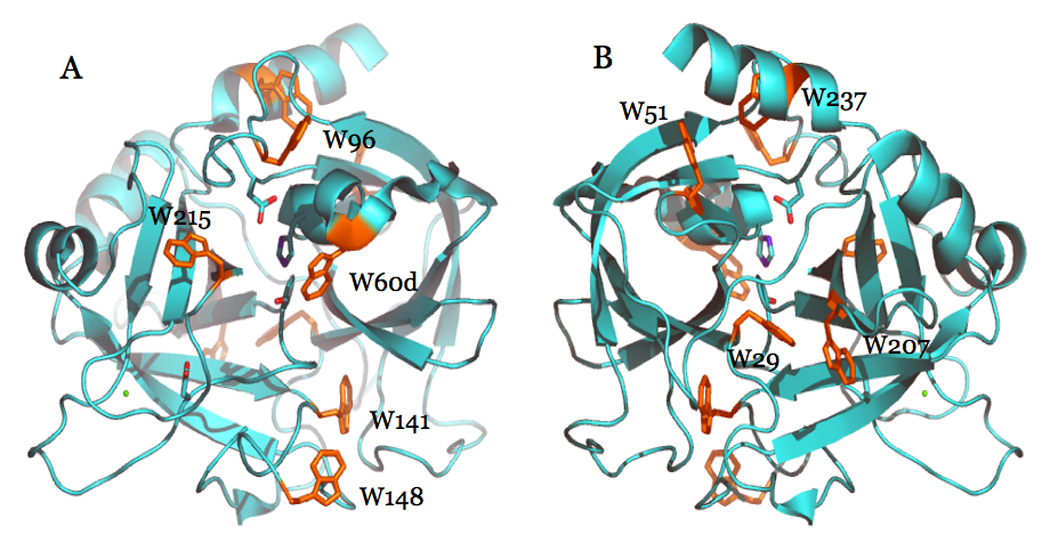
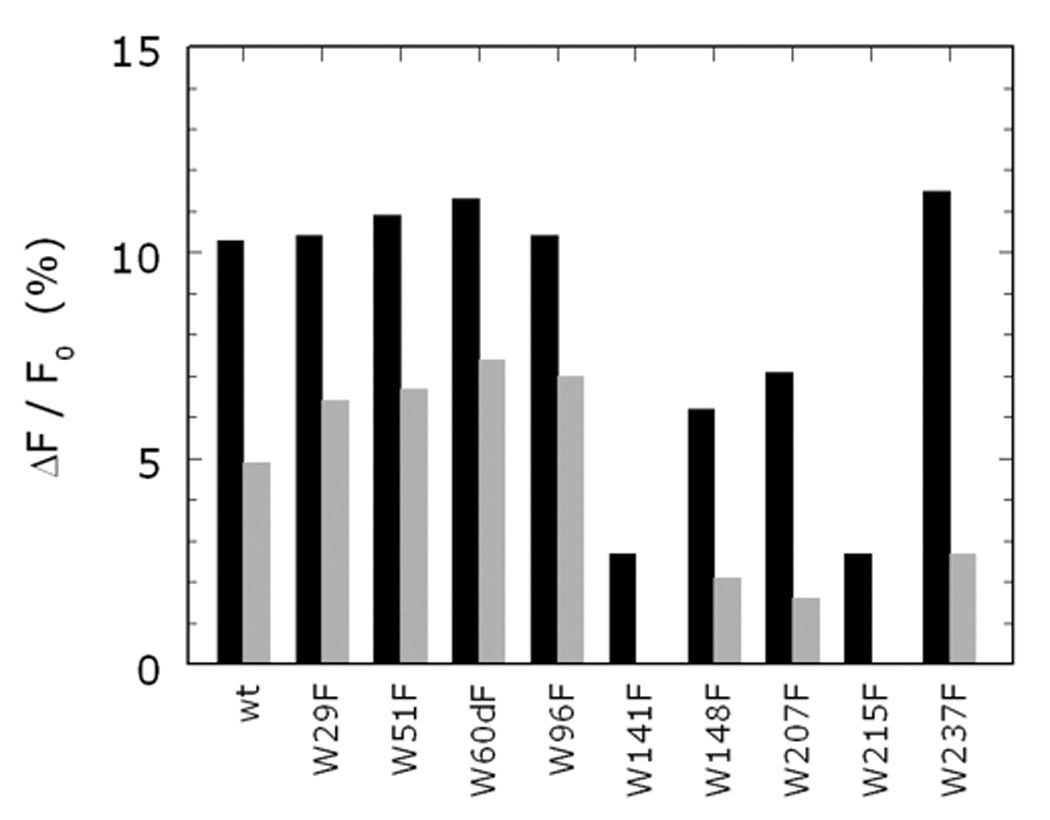
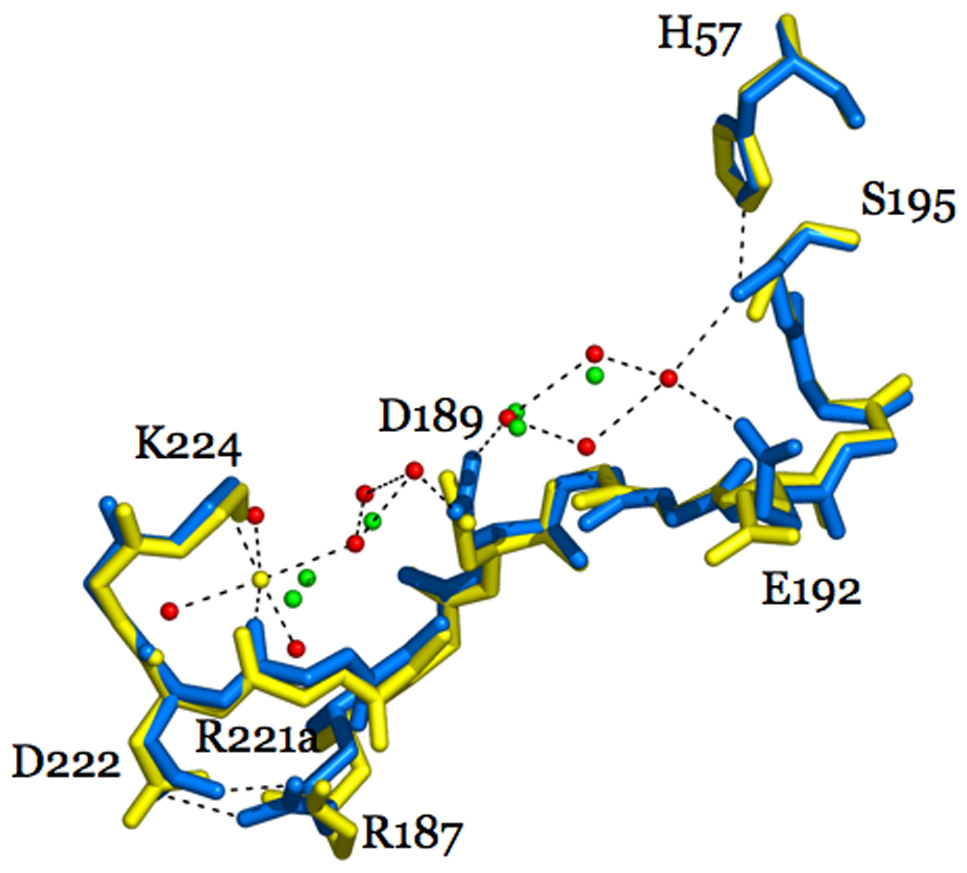
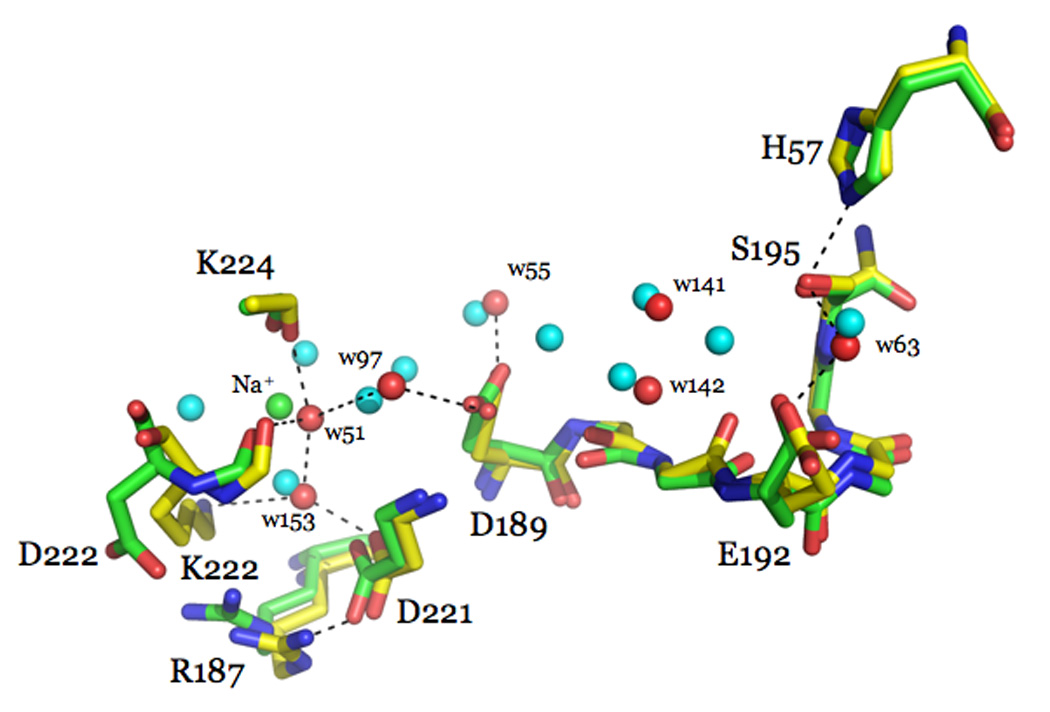
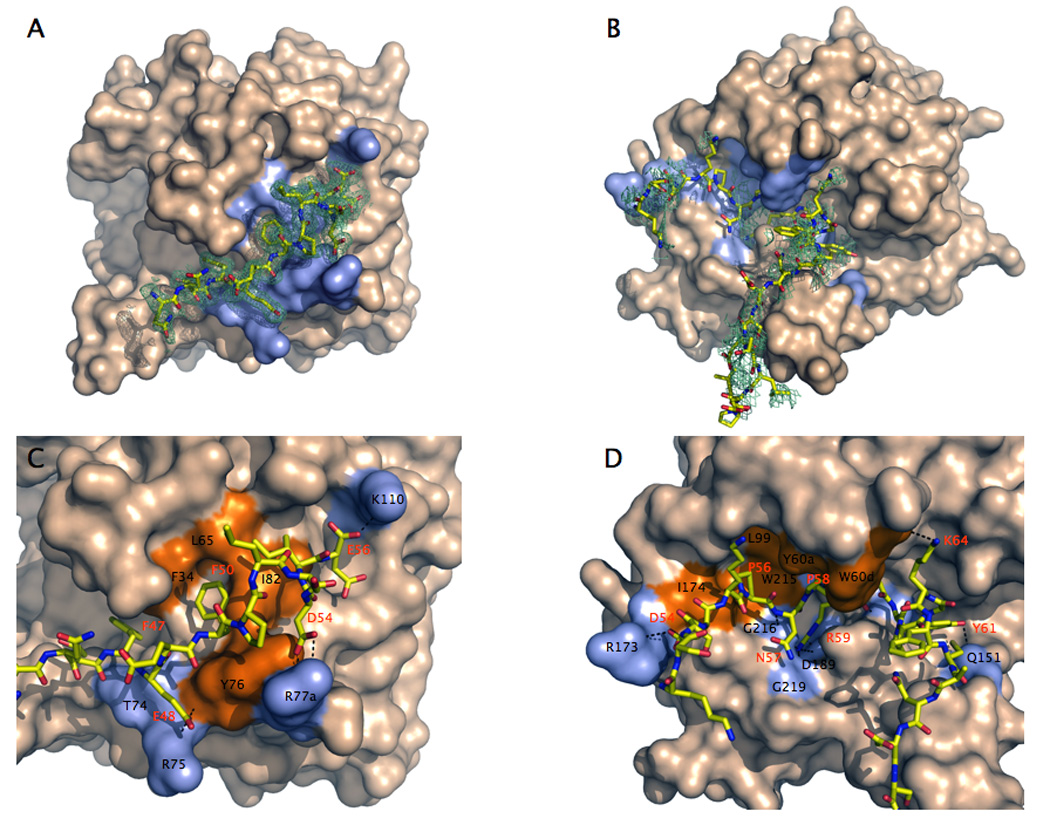
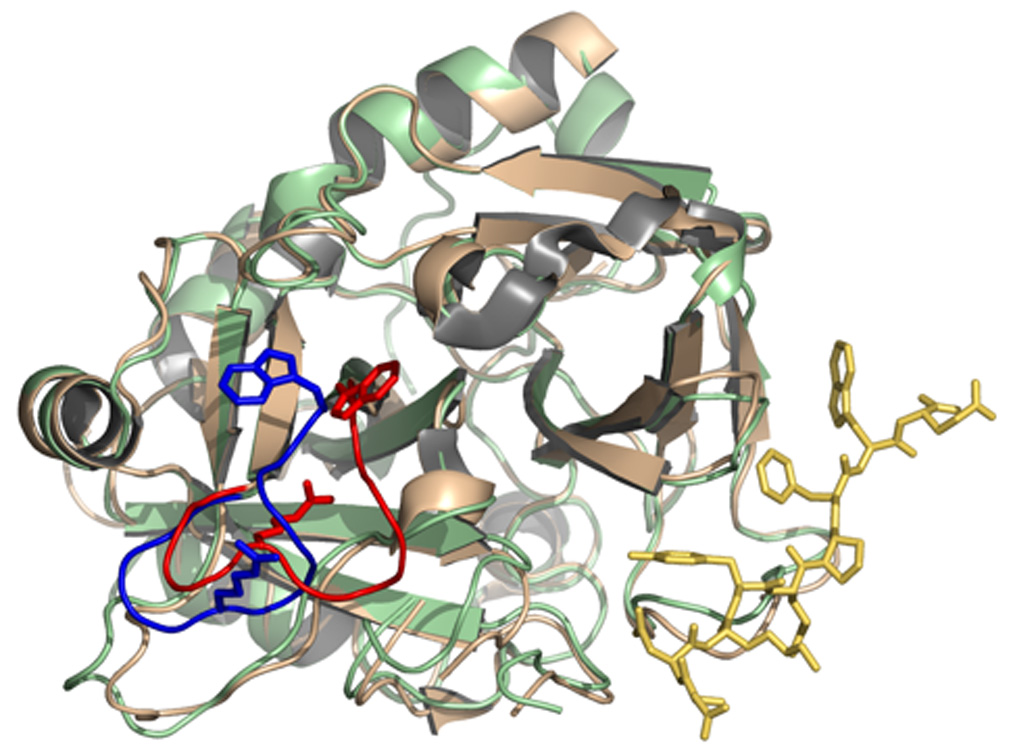
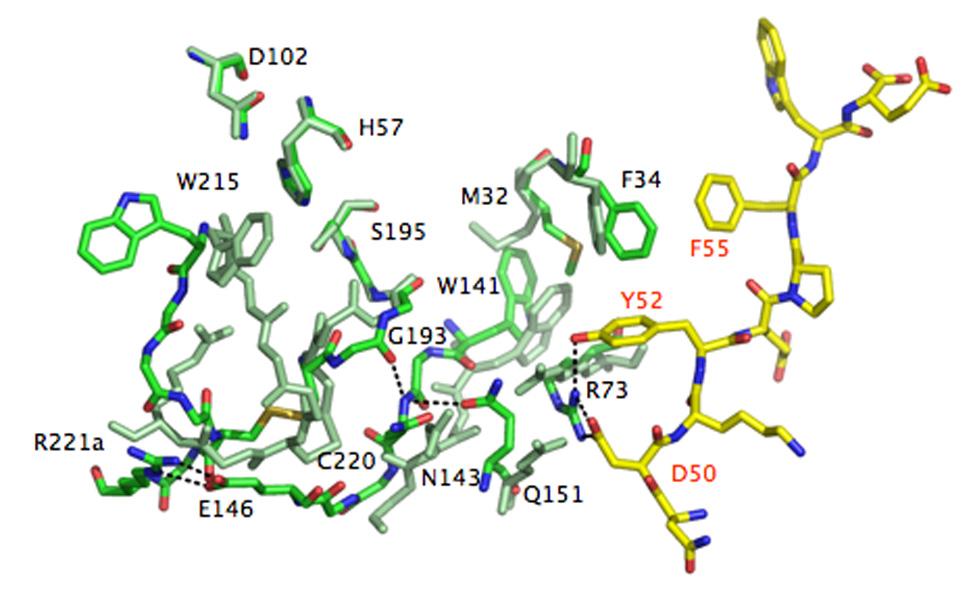
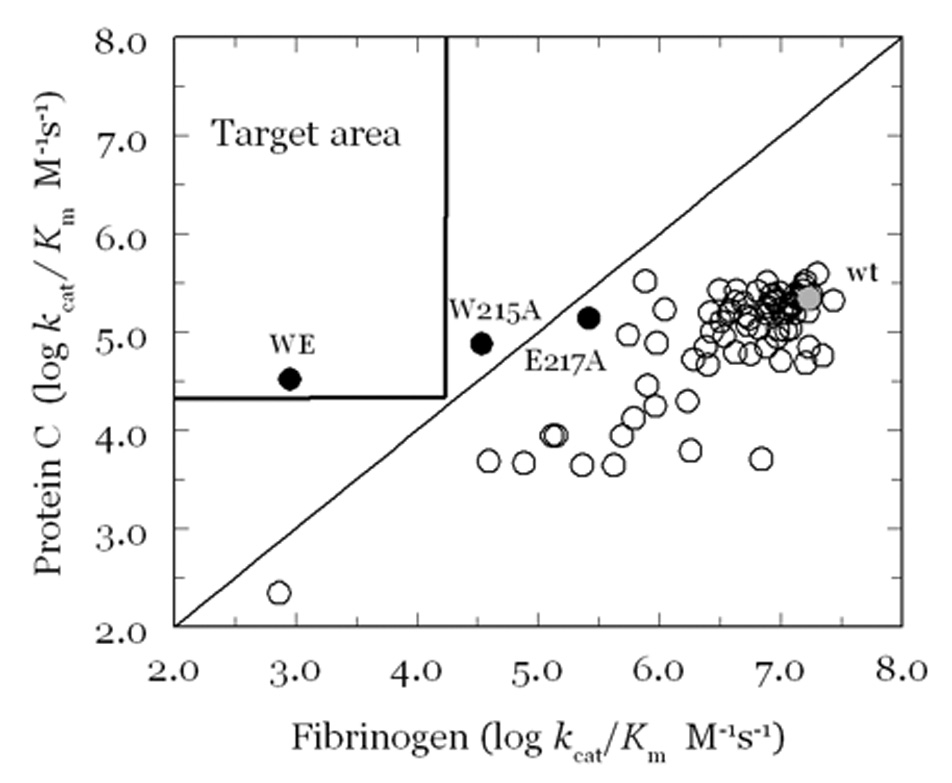
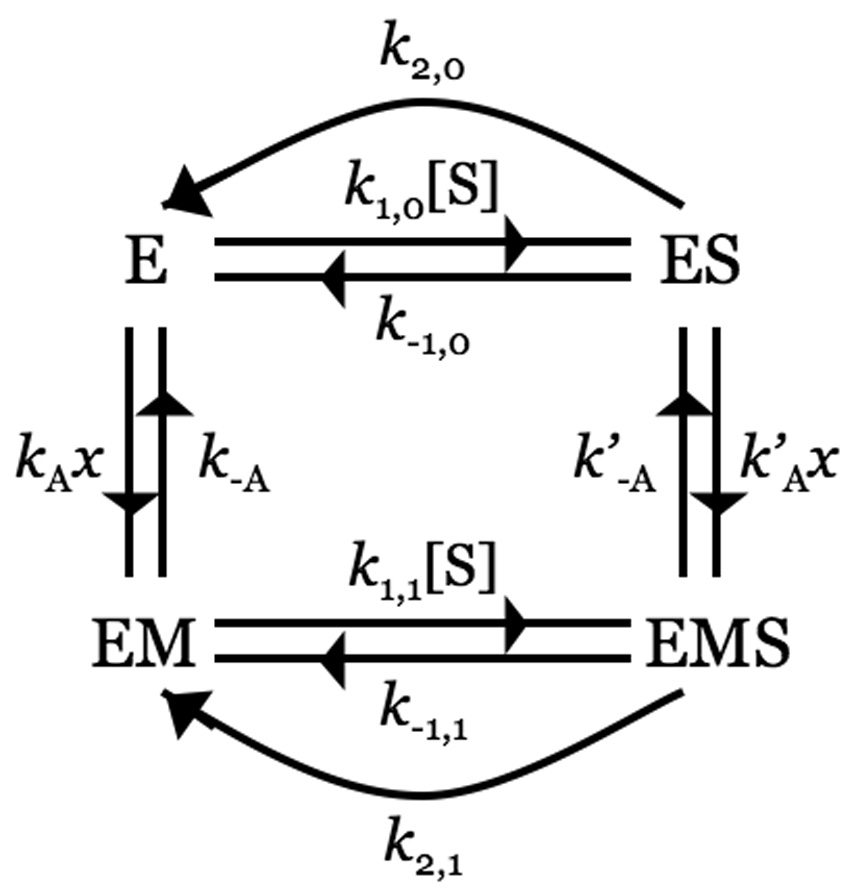
Thrombin is a Na+-activated, allosteric serine protease that plays opposing functional roles in blood coagulation. Binding of Na+ is the major driving force behind the procoagulant, prothrombotic and signaling functions of the enzyme, but is dispensable for cleavage of the anticoagulant protein C. The anticoagulant function of thrombin is under the allosteric control of the cofactor thrombomodulin. Much has been learned on the mechanism of Na+ binding and recognition of natural substrates by thrombin. Recent structural advances have shed light on the remarkable molecular plasticity of this enzyme and the molecular underpinnings of thrombin allostery mediated by binding to exosite I and the Na+ site. This review summarizes our current understanding of the molecular basis of thrombin function and allosteric regulation. The basic information emerging from recent structural, mutagenesis and kinetic investigation of this important enzyme is that thrombin exists in three forms, E*, E and E:Na+, that interconvert under the influence of ligand binding to distinct domains. The transition between the Na+ -free slow from E and the Na+ -bound fast form E:Na+ involves the structure of the enzyme as a whole, and so does the interconversion between the two Na+ -free forms E* and E. E* is most likely an inactive form of thrombin, unable to interact with Na + and substrate. The complexity of thrombin function and regulation has gained this enzyme pre-eminence as the prototypic allosteric serine protease. Thrombin is now looked upon as a model system for the quantitative analysis of biologically important enzymes.
Figures














References
-
- Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000a;342:1493–1499. - PubMed
-
- Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000b;342:1581–1589. - PubMed
-
- Akhavan S, Mannucci PM, Lak M, Mancuso G, Mazzucconi MG, Rocino A, Jenkins PV, Perkins SJ. Identification and three-dimensional structural analysis of nine novel mutations in patients with prothrombin deficiency. Thromb Haemost. 2000;84:989–997. - PubMed
-
- Akhavan S, Rocha E, Zeinali S, Mannucci PM. Gly319 --> arg substitution in the dysfunctional prothrombin Segovia. Br J Haematol. 1999;105:667–669. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
