

wuHMM: a robust algorithm to detect DNA copy number variation using long oligonucleotide microarray data
- PMID: 18334530
- PMCID: PMC2367727
- DOI: 10.1093/nar/gkn110
wuHMM: a robust algorithm to detect DNA copy number variation using long oligonucleotide microarray data
Abstract
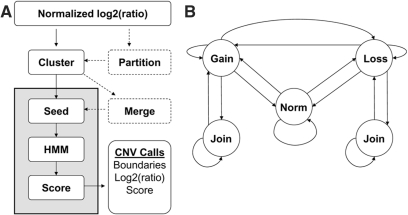
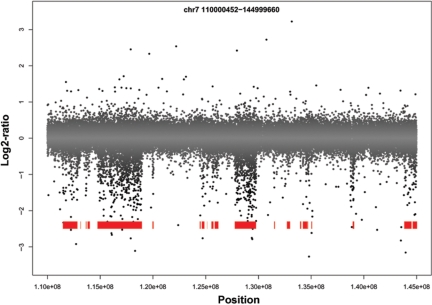
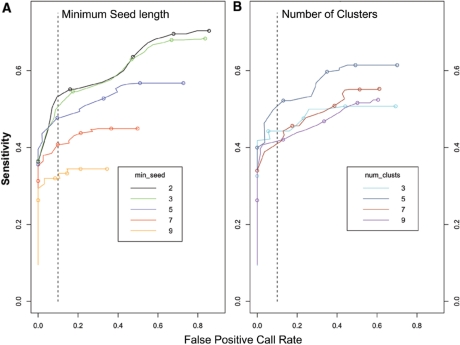
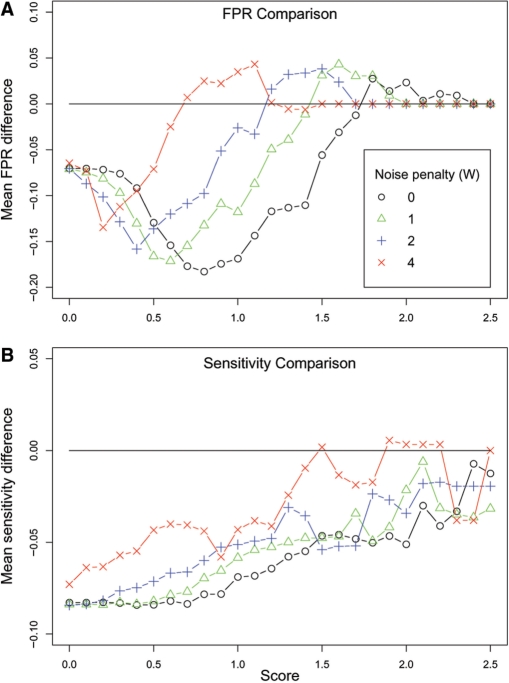
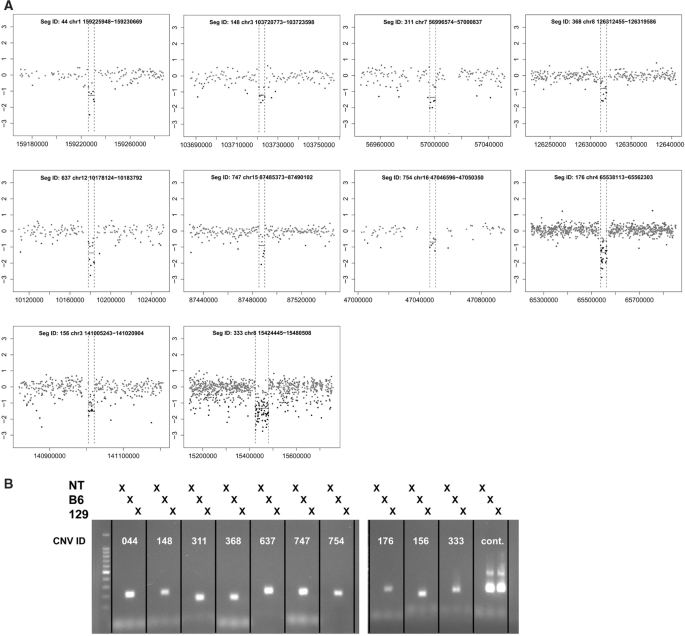
Copy number variants (CNVs) are currently defined as genomic sequences that are polymorphic in copy number and range in length from 1000 to several million base pairs. Among current array-based CNV detection platforms, long-oligonucleotide arrays promise the highest resolution. However, the performance of currently available analytical tools suffers when applied to these data because of the lower signal:noise ratio inherent in oligonucleotide-based hybridization assays. We have developed wuHMM, an algorithm for mapping CNVs from array comparative genomic hybridization (aCGH) platforms comprised of 385 000 to more than 3 million probes. wuHMM is unique in that it can utilize sequence divergence information to reduce the false positive rate (FPR). We apply wuHMM to 385K-aCGH, 2.1M-aCGH and 3.1M-aCGH experiments comparing the 129X1/SvJ and C57BL/6J inbred mouse genomes. We assess wuHMM's performance on the 385K platform by comparison to the higher resolution platforms and we independently validate 10 CNVs. The method requires no training data and is robust with respect to changes in algorithm parameters. At a FPR of <10%, the algorithm can detect CNVs with five probes on the 385K platform and three on the 2.1M and 3.1M platforms, resulting in effective resolutions of 24 kb, 2-5 kb and 1 kb, respectively.
Figures
References
-
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. - PubMed
-
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
