

IL-6/IL-6R axis plays a critical role in acute kidney injury
- PMID: 18337485
- PMCID: PMC2396933
- DOI: 10.1681/ASN.2007070744
IL-6/IL-6R axis plays a critical role in acute kidney injury
Abstract
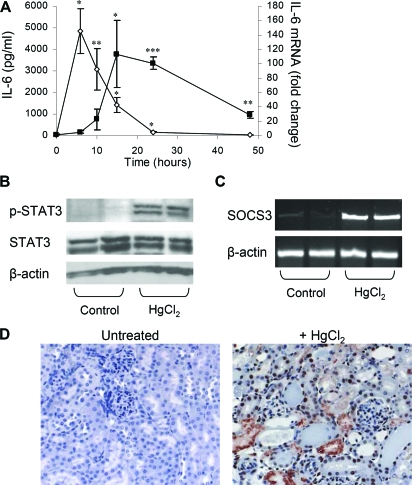
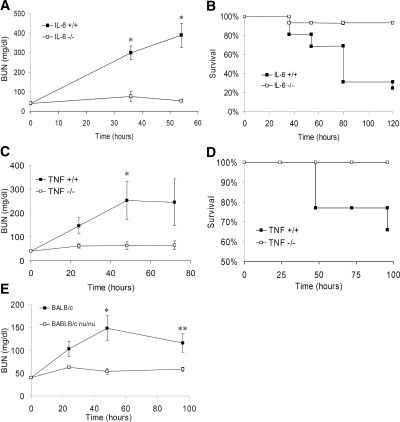
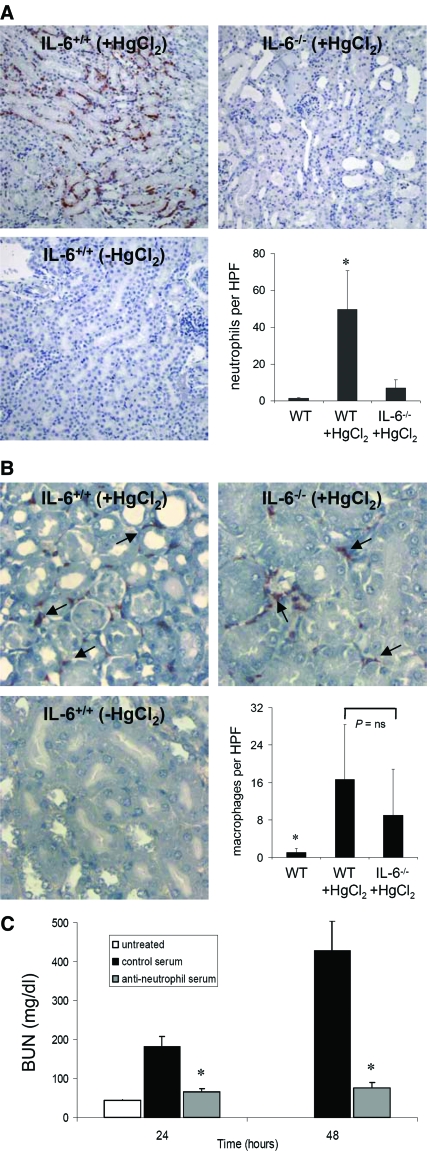
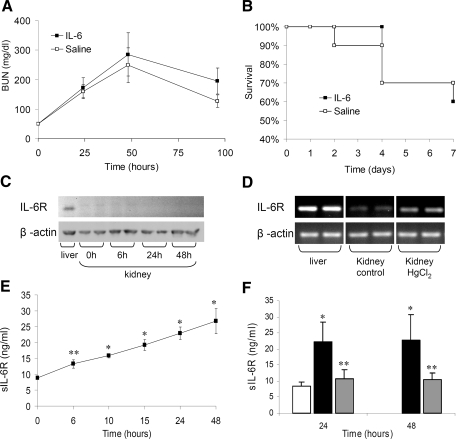
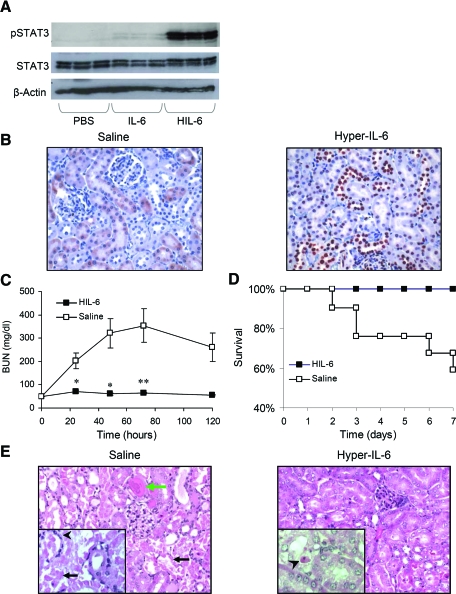
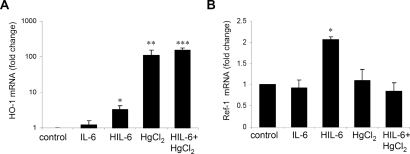
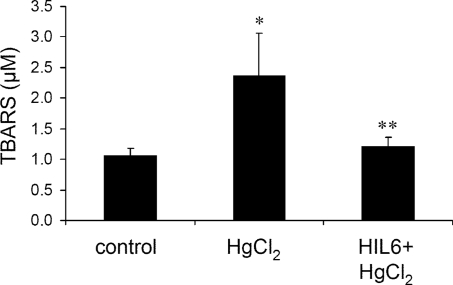
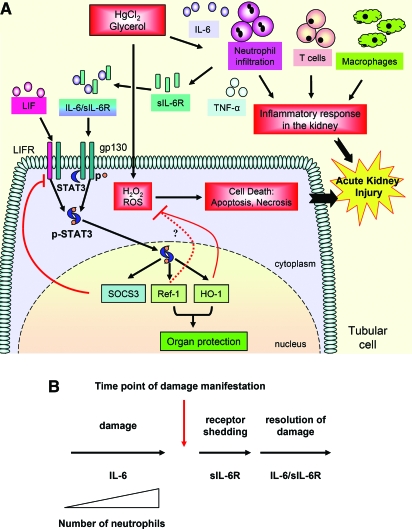
The response to tissue injury involves the coordination of inflammatory and repair processes. IL-6 expression correlates with the onset and severity of acute kidney injury (AKI), but its contribution to pathogenesis remains unclear. This study established a critical role for IL-6 in both the inflammatory response and the resolution of AKI. IL-6-deficient mice were resistant to HgCl2-induced AKI compared with wild-type mice. The accumulation of peritubular neutrophils was lower in IL-6-deficient mice than in wild-type mice, and neutrophil depletion before HgCl2 administration in wild-type mice significantly reduced AKI; these results demonstrate the critical role of IL-6 signaling in the injurious inflammatory process in AKI. Renal IL-6 expression and STAT3 activation in renal tubular epithelial cells significantly increased during the development of injury, suggesting active IL-6 signaling. Although a lack of renal IL-6 receptors (IL-6R) precludes the activation of classical signaling pathways, IL-6 can stimulate target cells together with a soluble form of the IL-6R (sIL-6R) in a process termed trans-signaling. During injury,serum sIL-6R levels increased three-fold, suggesting a possible role for IL-6 trans-signaling in AKI. Stimulation of IL-6 trans-signaling with an IL-6/sIL-6R fusion protein activated STAT3 in renal tubular epithelium and prevented AKI. IL-6/sIL-6R reduced lipid peroxidation after injury, suggesting that its protective effect may be largely mediated through amelioration of oxidative stress. In summary, IL-6 simultaneously promotes an injurious inflammatory response and, through a mechanism of trans-signaling, protects the kidney from further injury.
Figures
References
-
- Brady HR, Brenner BM: Acute renal failure. In: Harrison's Principles of Internal Medicine, 16th Ed., edited by Kasper DL, Fauci AS, Longo DL, Braunwald E, Hauser SL, Jameson JL, New York, McGraw-Hill, 2005, pp 1644–1653
-
- Simmons EM, Himmelfarb J, Sezer MT, Chertow GM, Mehta RL, Paganini EP, Soroko S, Freedman S, Becker K, Spratt D, Shyr Y, Ikizler TA: Plasma cytokine levels predict mortality in patients with acute renal failure. Kidney Int 65: 1357–1365, 2004 - PubMed
-
- Vaidya VS, Shankar K, Lock EA, Dixon D, Mehendale HM: Molecular mechanisms of renal tissue repair in survival from acute renal tubule necrosis: Role of ERK1/2 pathway. Toxicol Pathol 31: 604–618, 2003 - PubMed
-
- Lemay S, Rabb H, Postler G, Singh AK: Prominent and sustained up-regulation of gp130-signaling cytokines and the chemokine MIP-2 in murine renal ischemia-reperfusion injury. Transplantation 69: 959–963, 2000 - PubMed
-
- Gadient RA, Patterson PH: Leukemia inhibitory factor, Interleukin 6, and other cytokines using the GP130 transducing receptor: Roles in inflammation and injury. Stem Cells 17: 127–137, 1999 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
