

PPARdelta regulates multiple proinflammatory pathways to suppress atherosclerosis
- PMID: 18337509
- PMCID: PMC2393796
- DOI: 10.1073/pnas.0711875105
PPARdelta regulates multiple proinflammatory pathways to suppress atherosclerosis
Abstract
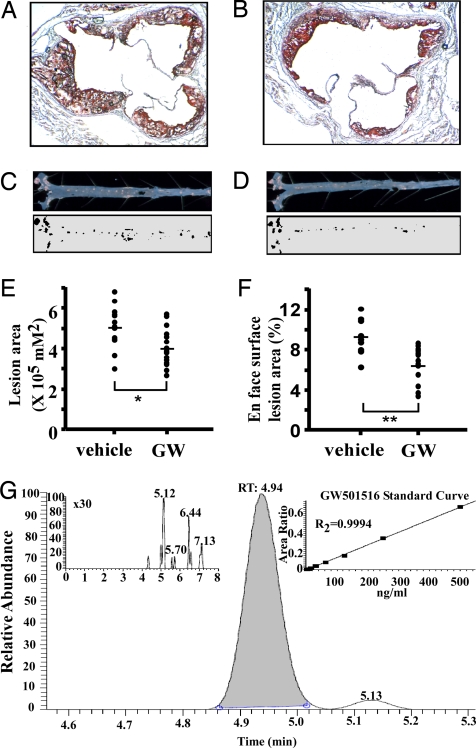
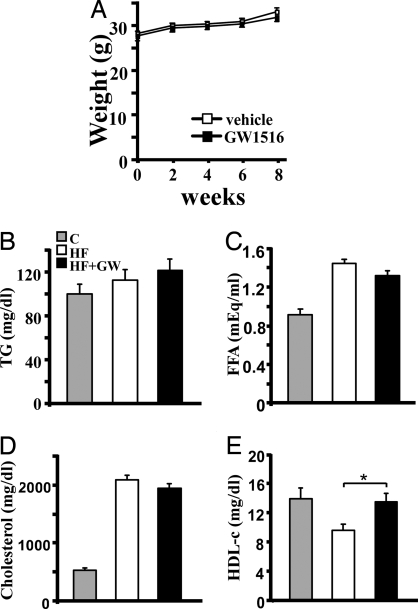
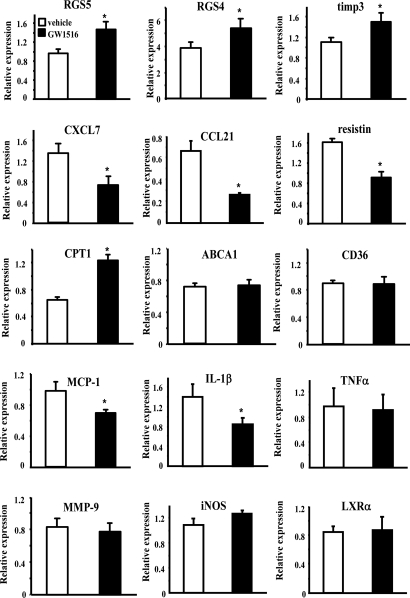
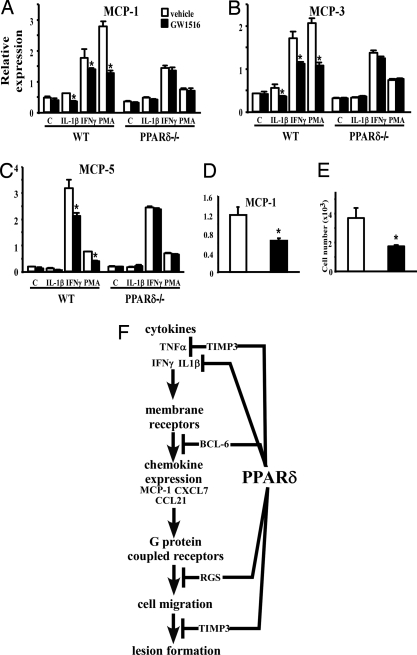
Lipid homeostasis and inflammation are key determinants in atherogenesis, exemplified by the requirement of lipid-laden, foam cell macrophages for atherosclerotic lesion formation. Although the nuclear receptor PPARdelta has been implicated in both systemic lipid metabolism and macrophage inflammation, its role as a therapeutic target in vascular disease is unclear. We show here that orally active PPARdelta agonists significantly reduce atherosclerosis in apoE(-/-) mice. Metabolic and gene expression studies reveal that PPARdelta attenuates lesion progression through its HDL-raising effect and anti-inflammatory activity within the vessel wall, where it suppresses chemoattractant signaling by down-regulation of chemokines. Activation of PPARdelta also induces the expression of regulator of G protein signaling (RGS) genes, which are implicated in blocking the signal transduction of chemokine receptors. Consistent with this, PPARdelta ligands repress monocyte transmigration and macrophage inflammatory responses elicited by atherogenic cytokines. These results reveal that PPARdelta antagonizes multiple proinflammatory pathways and suggest PPARdelta-selective drugs as candidate therapeutics for atherosclerosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104:503–516. - PubMed
-
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. - PubMed
-
- Gu L, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281. - PubMed
-
- Dawson TC, Kuziel WA, Osahar TA, Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
