

Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage?
- PMID: 18339586
- PMCID: PMC2408373
- DOI: 10.1016/j.dnarep.2008.01.017
Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage?
Abstract
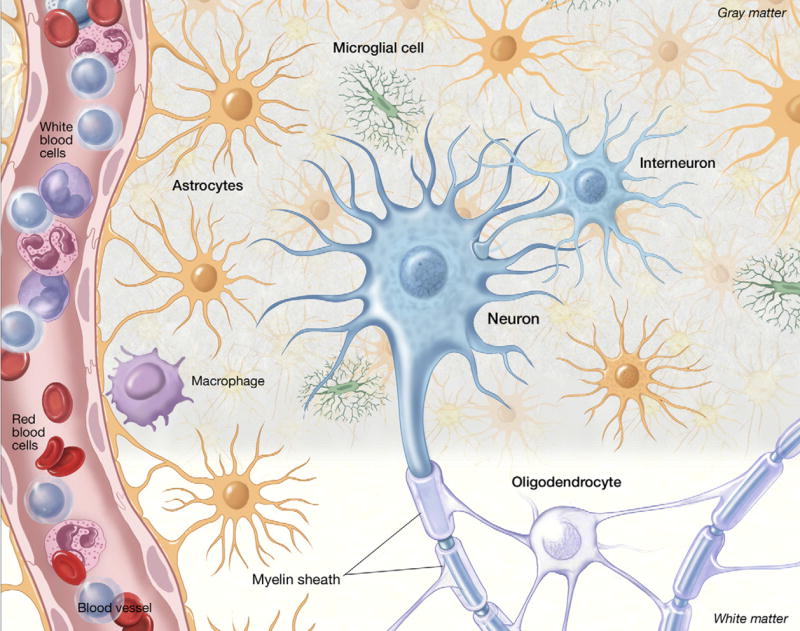
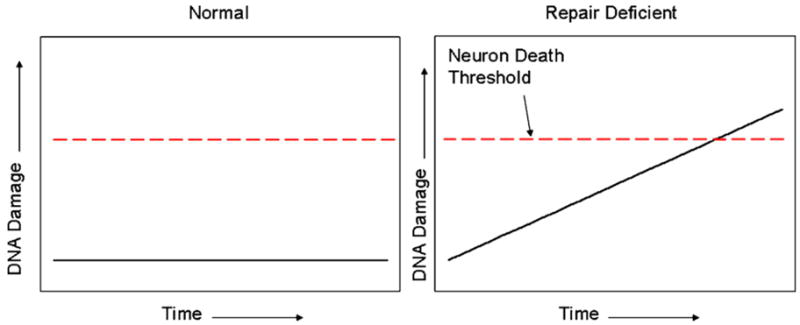
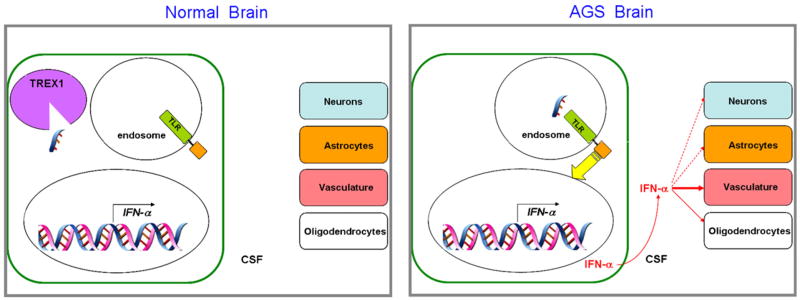
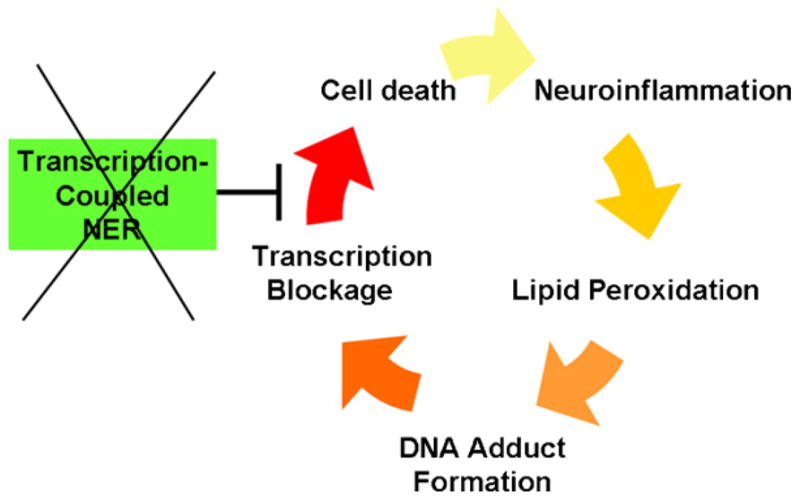
The classic model for neurodegeneration due to mutations in DNA repair genes holds that DNA damage accumulates in the absence of repair, resulting in the death of neurons. This model was originally put forth to explain the dramatic loss of neurons observed in patients with xeroderma pigmentosum neurologic disease, and is likely to be valid for other neurodegenerative diseases due to mutations in DNA repair genes. However, in trichiothiodystrophy (TTD), Aicardi-Goutières syndrome (AGS), and Cockayne syndrome (CS), abnormal myelin is the most prominent neuropathological feature. Myelin is synthesized by specific types of glial cells called oligodendrocytes. In this review, we focus on new studies that illustrate two disease mechanisms for myelin defects resulting from mutations in DNA repair genes, both of which are fundamentally different than the classic model described above. First, studies using the TTD mouse model indicate that TFIIH acts as a co-activator for thyroid hormone-dependent gene expression in the brain, and that a causative XPD mutation in TTD results in reduction of this co-activator function and a dysregulation of myelin-related gene expression. Second, in AGS, which is caused by mutations in either TREX1 or RNASEH2, recent evidence indicates that failure to degrade nucleic acids produced during S-phase triggers activation of the innate immune system, resulting in myelin defects and calcification of the brain. Strikingly, both myelin defects and brain calcification are both prominent features of CS neurologic disease. The similar neuropathology in CS and AGS seems unlikely to be due to the loss of a common DNA repair function, and based on the evidence in the literature, we propose that vascular abnormalities may be part of the mechanism that is common to both diseases. In summary, while the classic DNA damage accumulation model is applicable to the neuronal death due to defective DNA repair, the myelination defects and brain calcification seem to be better explained by quite different mechanisms. We discuss the implications of these different disease mechanisms for the rational development of treatments and therapies.
Figures
References
-
- Friedberg E, Walker G, Siede W, Wood R, Schultz R, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, DC: 2005.
-
- Kandel E, Schwatrz J, Jessel T. Principles of Neural Science. Elsevier; New York: 1991.
-
- Ledoux SP, Shen CC, Grishko VI, Fields PA, Gard AL, Wilson GL. Glial cell-specific differences in response to alkylation damage. Glia. 1998;24:304–312. - PubMed
-
- Hollensworth SB, Shen C, Sim JE, Spitz DR, Wilson GL, LeDoux SP. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic Biol Med. 2000;28:1161–1174. - PubMed
-
- Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–397. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
