

doi: 10.1038/nchembio0408-217.
Evolving biosynthetic tangos negotiate mechanistic landscapes
Affiliations
- PMID: 18347585
- PMCID: PMC2703434
- DOI: 10.1038/nchembio0408-217
Item in Clipboard
Evolving biosynthetic tangos negotiate mechanistic landscapes
Nat Chem Biol.
2008 Apr.
Abstract
The dependence of polyketide synthase and terpene cyclase mechanistic adaptation on the chemistry of their oligomeric substrates illuminates a convergent evolutionary strategy for shaping cyclization in these otherwise disparate reactions. Evolution of these enzyme families relies on rhythmic tangos, in which the enzymes and substrates together determine product outcome by negotiating decision networks governing intrinsic and induced chemical reactivities.
Figures
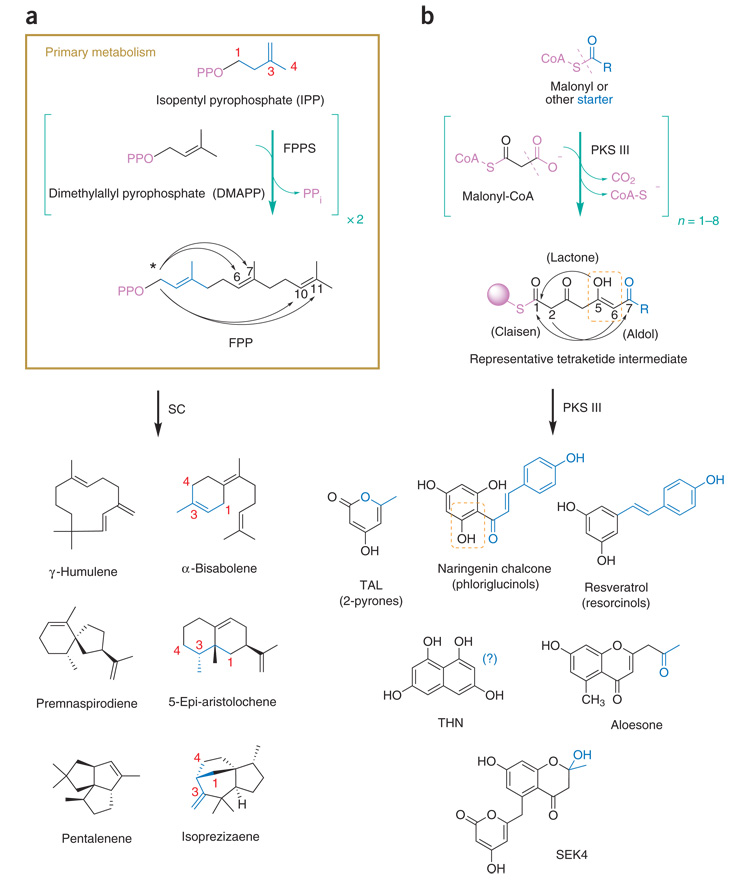
Metabolic diversity via divergent intramolecular cyclization. Building blocks, linear oligomeric intermediates and representative uni-, bi- and tricyclic products biosynthesized by two cyclase families are shown. Leaving groups (violet) are exploited for enzyme recognition and reactivity enhancement. Curved arrows indicate alternate initial intramolecular condensations. (a) SCs cyclize FPP, an isoprenoid trimer from primary metabolism, to biosynthesize hundreds of distinct cyclic sesquiterpene hydrocarbons. Some initial intramolecular condensations of FPP-derived carbocations require a prior isomerization event to a cisoid farnesyl cation (arrows marked by *), whereas others proceed by direct attack of the nascent carbocation following ionization. A highlighted isoprene unit (blue) reveals additional rearrangements en route to SC products (see text). (b) PKS IIIs selectively transfer a CoA-thioester–activated starter moiety (blue) to a catalytic cysteine, catalyze iterative polyketide chain extension via decarboxylative condensations with malonyl-CoA and then divergently cyclize the resulting linear oligomeric intermediates to offload products. Subunits of extended polyketide intermediates can undergo keto-enol tautomerization (dashed orange box). Note that the internal symmetry of the THN product obscures the final position of the incorporated starter moiety (blue).
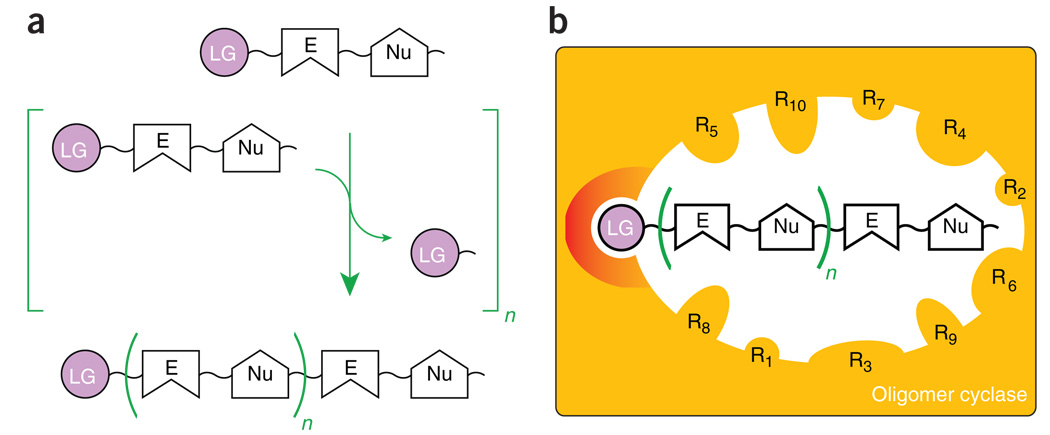
Convergent features of ‘uber-adaptable’ cyclases and their oligomeric substrates. (a) Polymerization involves covalent bond formation between a nucleophile (Nu) and an electrophile (E). Enzymes exploit high-energy bonds tethered to favorable leaving groups (LG) to activate adjacent electrophiles. These moieties also provide convenient chemical handles for modular small-molecule recognition. (b) A generic cyclase is depicted as an orange box. Structural analyses of unrelated PKS IIIs and SCs reveal analogous internal active site cavities lined by amino acid side chains (R), allowing evolving steric control over oligomeric substrate/intermediate conformations leading to alternative juxtapositions of the repetitive and reactive Nu-E pairs. Evolutionary conservation of catalytic machinery (red) for binding and manipulating LGs is also shown.
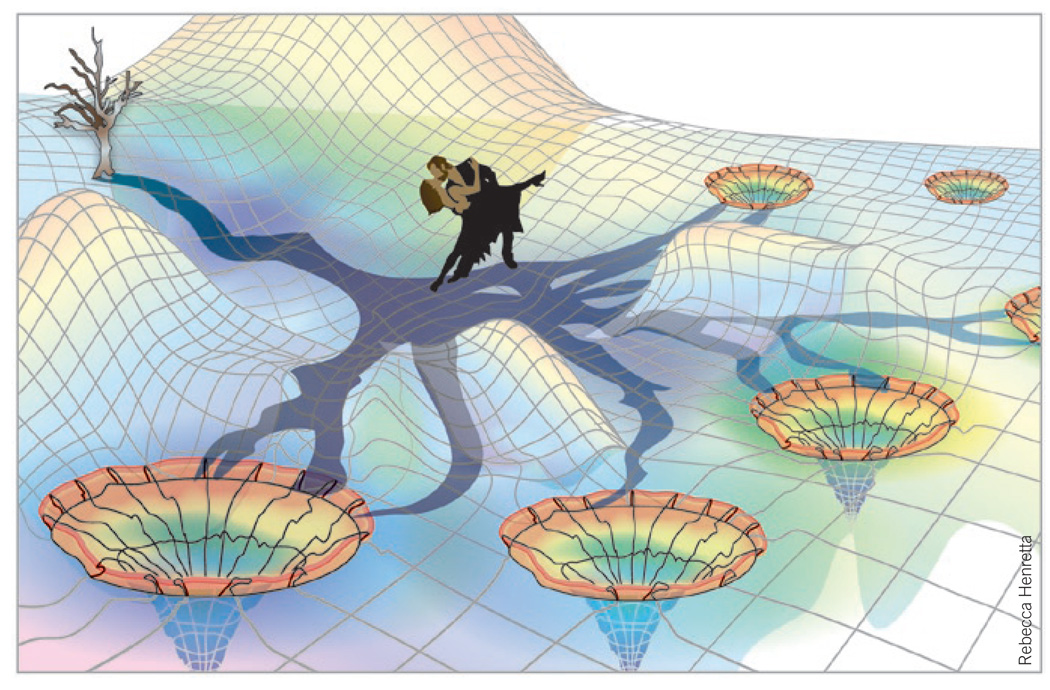
Biosynthetic tangos in the shade of a branching metabolic diversification tree. The vertical tree represents the accessible product spectrum of a particular cyclase-substrate dance pair and is rooted at the edge of the (enzyme-bound) small-molecule conformational energy landscape. The tree casts its shadow across this uneven dance floor, thus highlighting a corresponding horizontal mechanistic decision network. Multistep negotiation of this network across the dance floor constitutes the ‘biosynthetic tango’ between substrate and enzyme that determines the accessibility of alternative reaction pathways leading to distinct stable products.
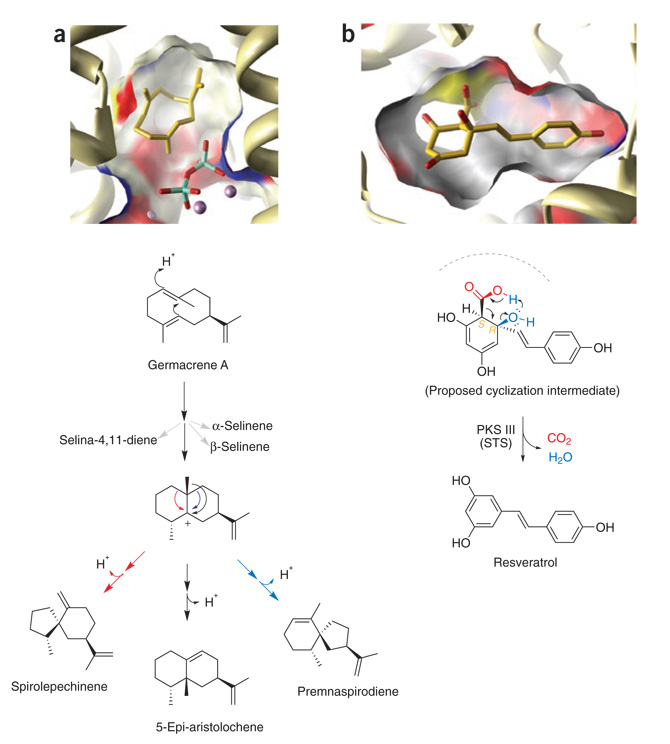
Comparison of postcyclization reactions in the SC (left) and PKS III (right) ‘reaction chambers’. Transient intermediates (gold stick models) are modeled in cut-away surface views of the active site cavities (colored by atom type) and surrounding protein (ribbons). (a) Divergent TEAS-catalyzed cyclization of the neutral germacrene A intermediate. Following Mg2+-assisted elimination of PPi (bottom right of active site figure), TEAS forms germacrene A from the initial C1,10 cyclization and deprotonation of the farnesyl cation (see Figure 1). Protonation of germacrene A regenerates a carbocation to launch a new series of electrophilic cyclizations marked by competing alkyl shifts and terminating in alternative quenching via proton elimination. (b) Rapid collapse to aromaticity obscures the intervening stereochemistry of PKS III cyclization intermediates. Modeled is an unstable and proposed cyclic intermediate of the stilbene synthase (STS)-catalyzed tetraketide aldol condensation leading to resveratrol (see Figure 1), viewed from the perspective of the ‘aldol switch’. Also visible are the catalytic cysteine (yellow surface) and the CoA-binding tunnel. Shown below, the coupled elimination of a carboxylate (red) and adjacent hydroxyl group (blue) following the STS-catalyzed aldol condensation reaction.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
