

Comparing whole genomes using DNA microarrays
- PMID: 18347592
- PMCID: PMC7097741
- DOI: 10.1038/nrg2335
Comparing whole genomes using DNA microarrays
Abstract
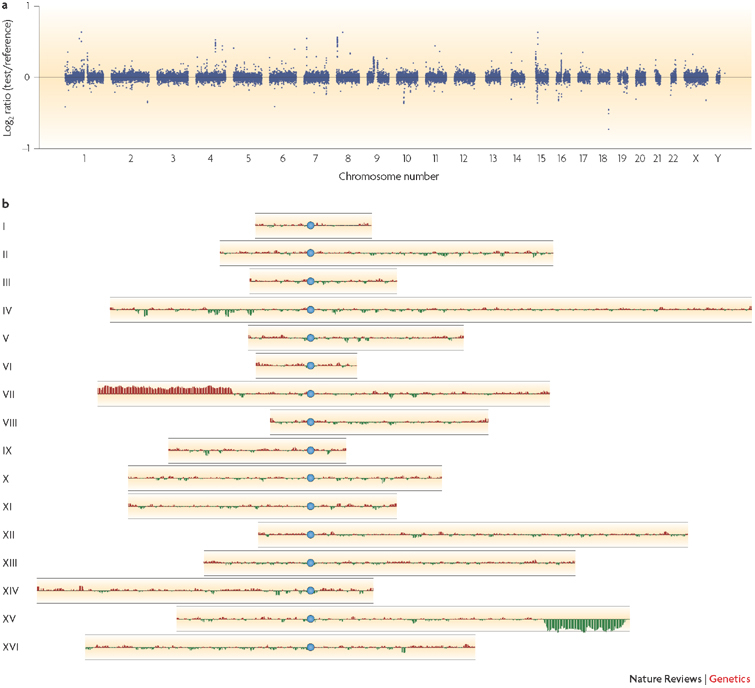
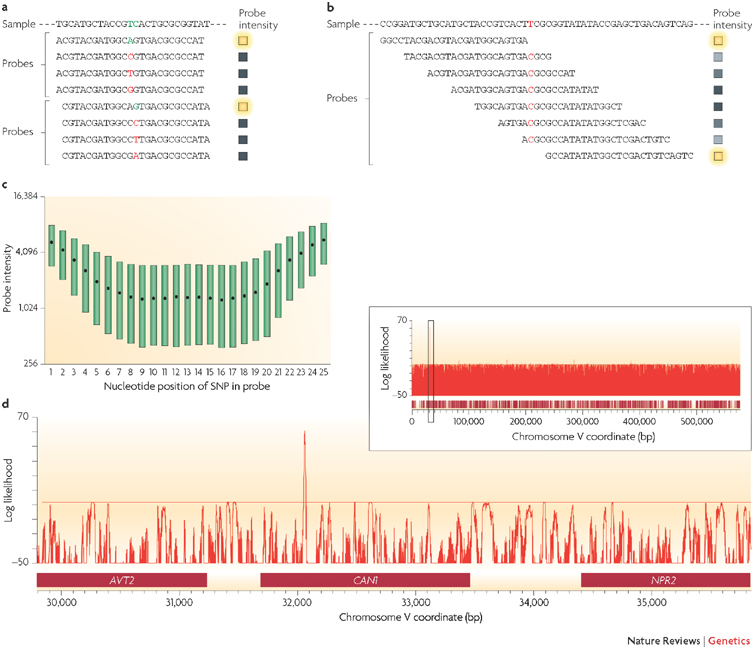
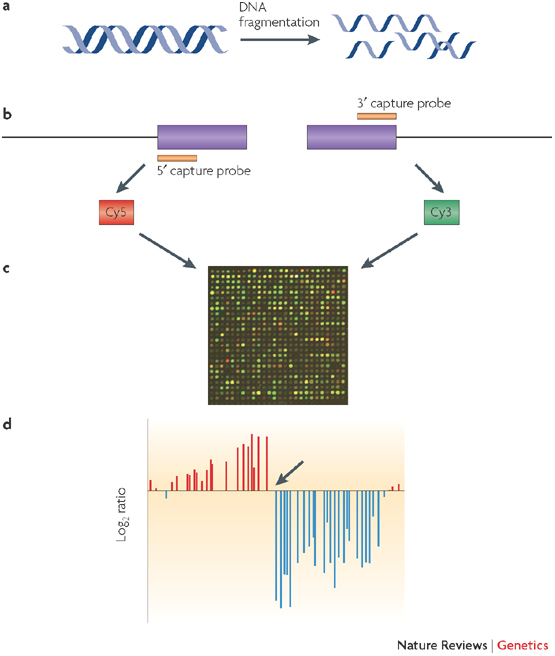
The rapid accumulation of complete genomic sequences offers the opportunity to carry out an analysis of inter- and intra-individual genome variation within a species on a routine basis. Sequencing whole genomes requires resources that are currently beyond those of a single laboratory and therefore it is not a practical approach for resequencing hundreds of individual genomes. DNA microarrays present an alternative way to study differences between closely related genomes. Advances in microarray-based approaches have enabled the main forms of genomic variation (amplifications, deletions, insertions, rearrangements and base-pair changes) to be detected using techniques that are readily performed in individual laboratories using simple experimental approaches.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
