

Differential regulation of the IL-17 receptor by gammac cytokines: inhibitory signaling by the phosphatidylinositol 3-kinase pathway
- PMID: 18348982
- PMCID: PMC2376247
- DOI: 10.1074/jbc.M801357200
Differential regulation of the IL-17 receptor by gammac cytokines: inhibitory signaling by the phosphatidylinositol 3-kinase pathway
Abstract
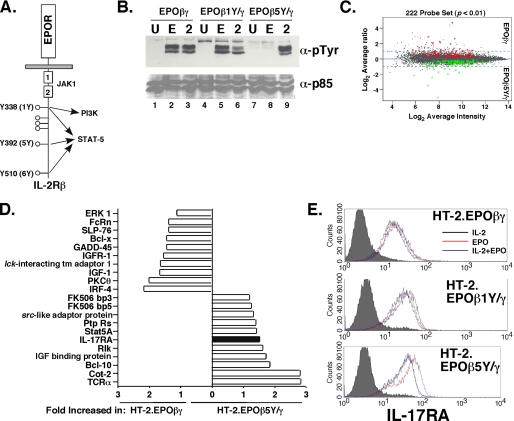
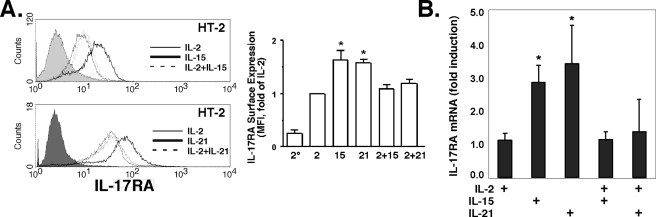
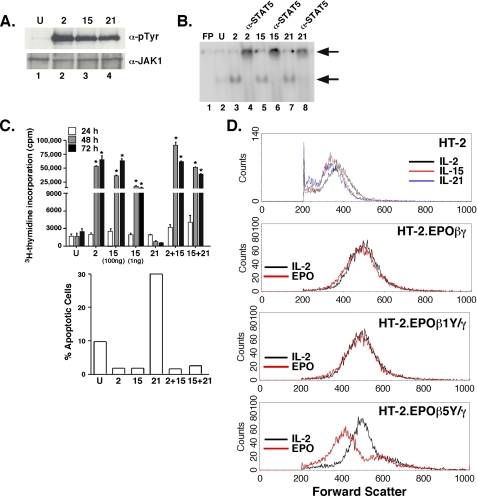
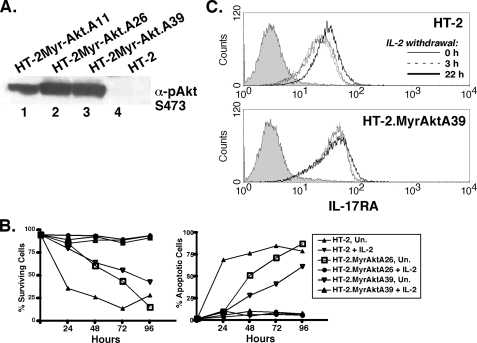
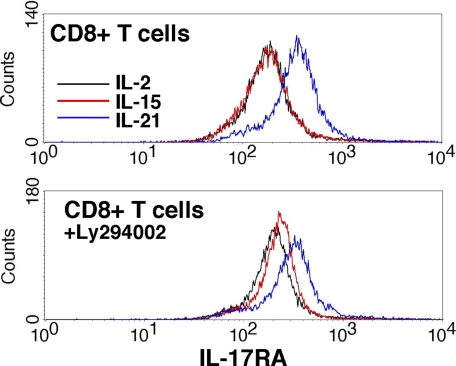
The gammac-family cytokine IL-2 activates signaling events that contribute to cell survival and proliferation, the best-studied of which are the STAT-5 and phosphatidylinositol 3-kinase (PI3K) pathways. The starting point of this study was to define genes regulated by the IL-2R-mediated PI3K pathway in T cells. Accordingly, we used an erythropoietin (EPO) receptor chimeric receptor system in which IL-2-dependent HT-2 T cells expressed a mutant EPO-IL-2Rbeta construct where Tyr-338 is mutated to Phe. Cells expressing this mutant IL-2Rbeta chain fail to induce phosphorylation of PI3K-p85alpha/beta or activate Akt, but mediate normal IL-2-dependent proliferation and activation of JAK1 and STAT-5A/B. Microarray analyses revealed differential regulation of numerous genes compared with cells expressing a wild-type IL-2Rbeta, including up-regulation of the IL-17 receptor subunit IL-17RA. Blockade of the PI3K pathway but not p70S6K led to up-regulation of IL-17RA, and constitutive Akt activation was associated with suppressed IL-17RA expression. Moreover, similar to the mutant EPO-IL-2Rbeta chimera, IL-15 and IL-21 induced IL-17RA preferentially compared with IL-2, and IL-2 but not IL-15 or IL-21 mediated prolonged activation of the PI3K p85 regulatory subunit. Thus, there are intrinsic signaling differences between IL-2 and IL-15 that can be attributed to differences in activation of the PI3K pathway.
Figures


References
-
- Ozaki, K., and Leonard, W. J. (2002) J. Biol. Chem. 277 29355-29358 - PubMed
-
- Ma, A. (2000) Mod. Asp. Immunobiol. 1 102-104
-
- Waldmann, T. (2006) Nat. Rev. 6 595-601 - PubMed
-
- Johnston, J. A., Kawamura, M., Kirken, R. A., Chen, Y. Q., Blake, T. B., Shibuya, K., Ortaldo, J. R., McVicar, D. W., and O'Shea, J. J. (1994) Nature 370 151-153 - PubMed
-
- Russell, S. M., Johnston, J. A., Noguchi, M., Kawamura, M., Bacon, C. M., Friedmann, M., Berg, M., McVicar, D. W., Whitthuhn, B. A., Silvennoinen, O., Goldman, A. S., Schmalsteig, F. C., Ihle, J. N., O'Shea, J. J., and Leonard, W. J. (1994) Science 266 1042-1045 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
