

Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels
- PMID: 18350175
- PMCID: PMC2267038
- DOI: 10.1371/journal.pone.0001852
Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels
Abstract
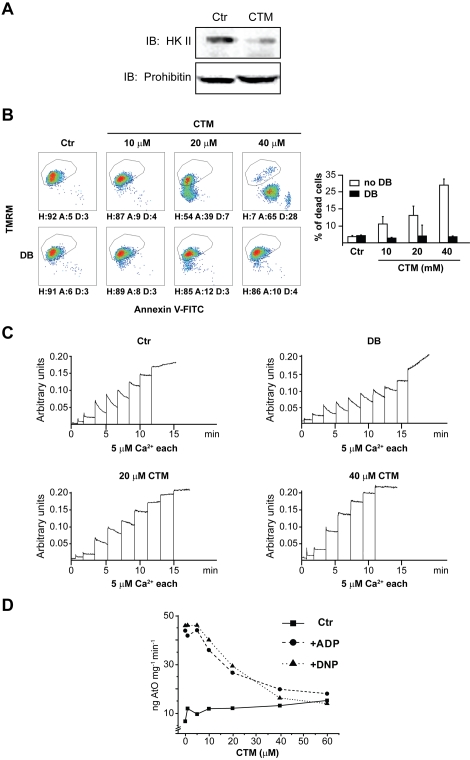
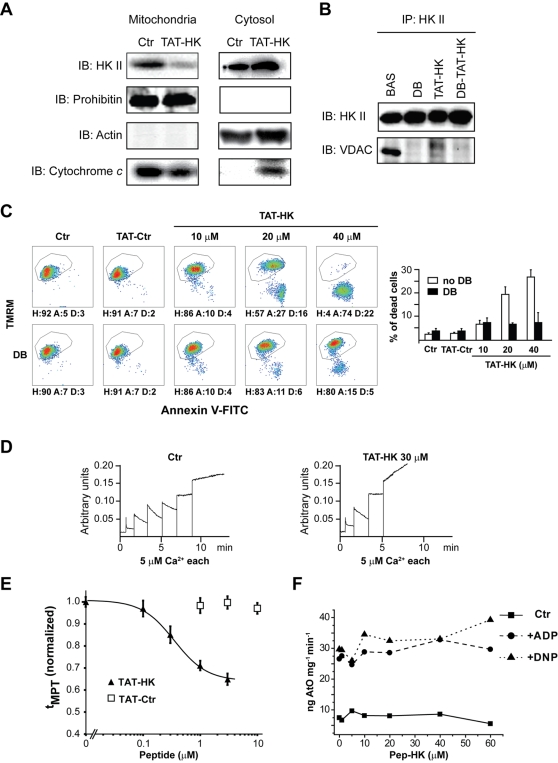
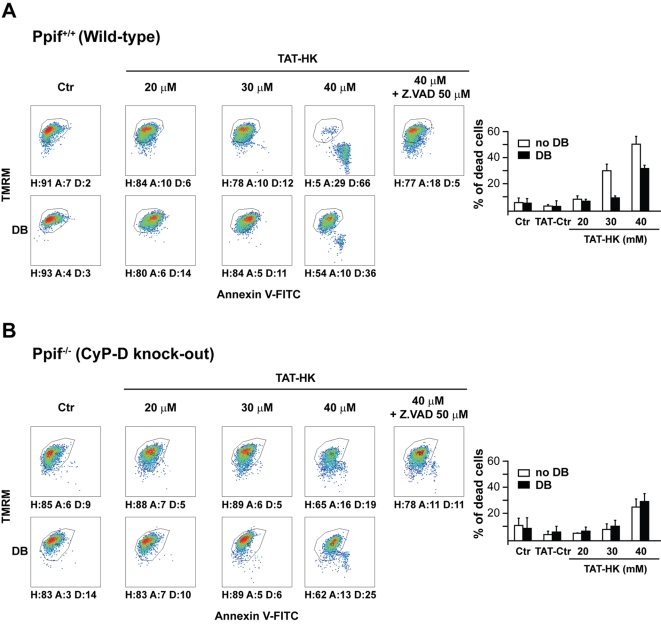
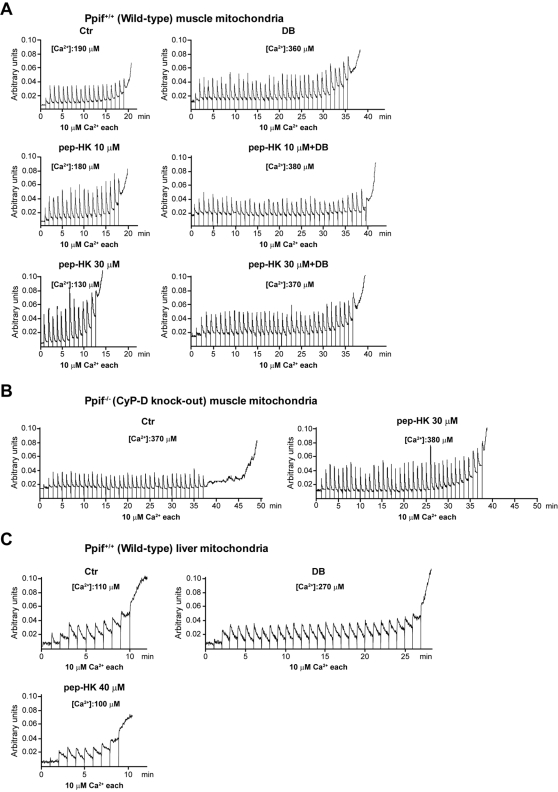
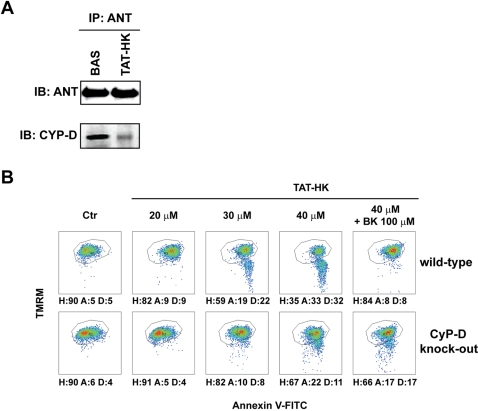
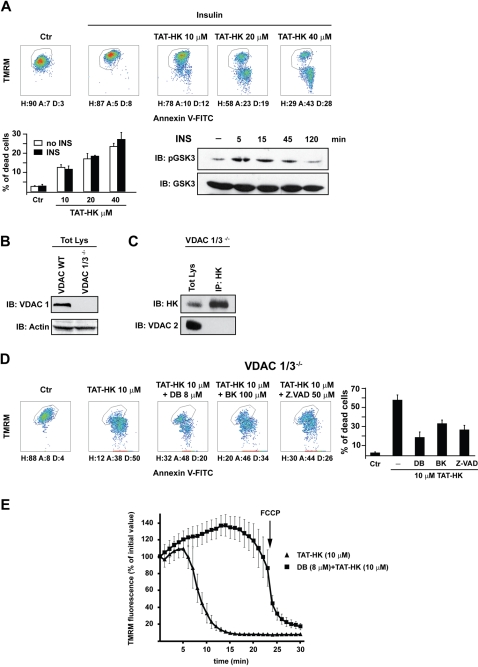
Type II hexokinase is overexpressed in most neoplastic cells, and it mainly localizes on the outer mitochondrial membrane. Hexokinase II dissociation from mitochondria triggers apoptosis. The prevailing model postulates that hexokinase II release from its mitochondrial interactor, the voltage-dependent anion channel, prompts outer mitochondrial membrane permeabilization and the ensuing release of apoptogenic proteins, and that these events are inhibited by growth factor signalling. Here we show that a hexokinase II N-terminal peptide selectively detaches hexokinase II from mitochondria and activates apoptosis. These events are abrogated by inhibiting two established permeability transition pore modulators, the adenine nucleotide translocator or cyclophilin D, or in cyclophilin D knock-out cells. Conversely, insulin stimulation or genetic ablation of the voltage-dependent anion channel do not affect cell death induction by the hexokinase II peptide. Therefore, hexokinase II detachment from mitochondria transduces a permeability transition pore opening signal that results in cell death and does not require the voltage-dependent anion channel. These findings have profound implications for our understanding of the pathways of outer mitochondrial membrane permeabilization and their inactivation in tumors.
Conflict of interest statement
Figures
References
-
- Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. - PubMed
-
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. - PubMed
-
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
