

Cytosine methylation profiling of cancer cell lines
- PMID: 18353987
- PMCID: PMC2290817
- DOI: 10.1073/pnas.0712251105
Cytosine methylation profiling of cancer cell lines
Abstract
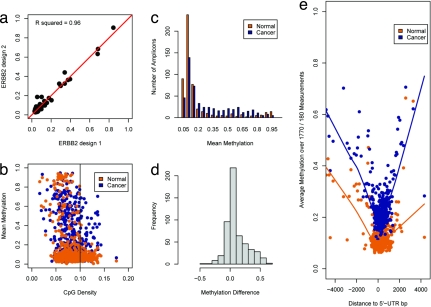
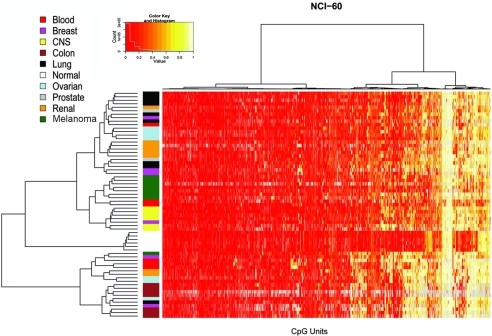
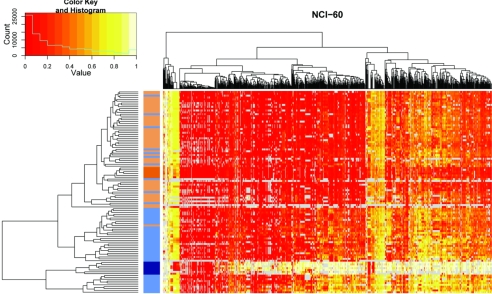
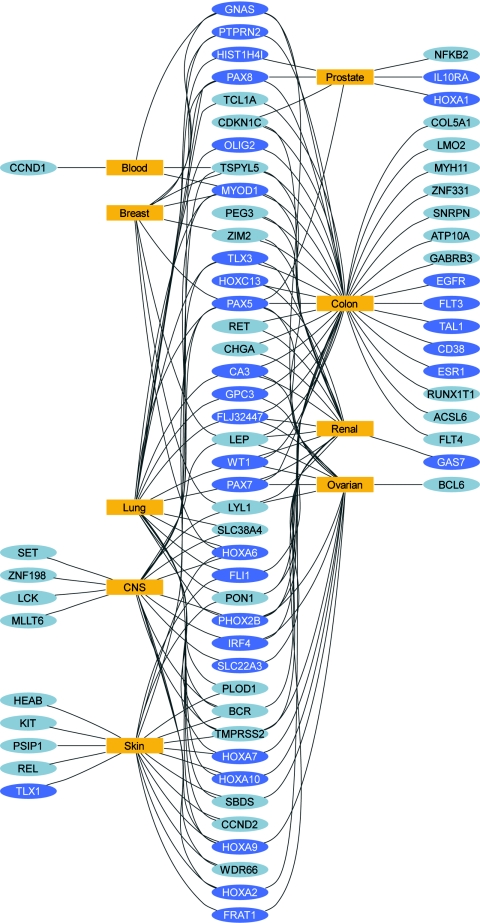
DNA-methylation changes in human cancer are complex and vary between the different types of cancer. Capturing this epigenetic variability in an atlas of DNA-methylation changes will be beneficial for basic research as well as translational medicine. Hypothesis-free approaches that interrogate methylation patterns genome-wide have already generated promising results. However, these methods are still limited by their quantitative accuracy and the number of CpG sites that can be assessed individually. Here, we use a unique approach to measure quantitative methylation patterns in a set of >400 candidate genes. In this high-resolution study, we employed a cell-line model consisting of 59 cancer cell lines provided by the National Cancer Institute and six healthy control tissues for discovery of methylation differences in cancer-related genes. To assess the effect of cell culturing, we validated the results from colon cancer cell lines by using clinical colon cancer specimens. Our results show that a large proportion of genes (78 of 400 genes) are epigenetically altered in cancer. Although most genes show methylation changes in only one tumor type (35 genes), we also found a set of genes that changed in many different forms of cancer (seven genes). This dataset can easily be expanded to develop a more comprehensive and ultimately complete map of quantitative methylation changes. Our methylation data also provide an ideal starting point for further translational research where the results can be combined with existing large-scale datasets to develop an approach that integrates epigenetic, transcriptional, and mutational findings.
Conflict of interest statement
Conflict of interest statement: M.E., J.T., C.C., and D.v.d.B. are shareholders and full-time employees of Sequenom, Inc.
Figures
References
-
- Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–266. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
