

Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer's disease
- PMID: 18363841
- PMCID: PMC4401126
- DOI: 10.1111/j.1582-4934.2008.00314.x
Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer's disease
Abstract
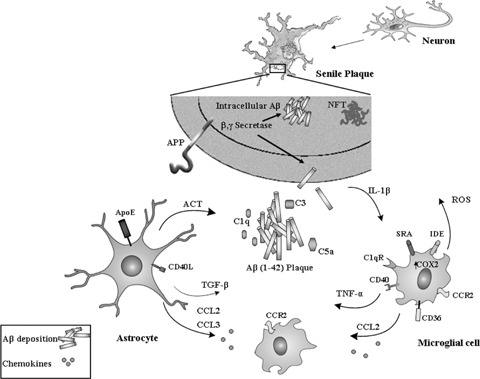
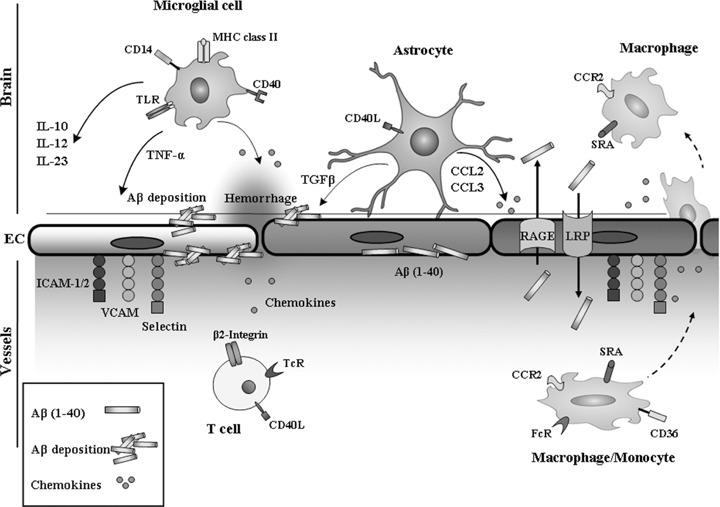
Alzheimer's disease (AD) affects more than 18 million people worldwide and is characterized by progressive memory deficits, cognitive impairment and personality changes. The main cause of AD is generally attributed to the increased production and accumulation of amyloid-beta (Abeta), in association with neurofibrillary tangle (NFT) formation. Increased levels of pro-inflammatory factors such as cytokines and chemokines, and the activation of the complement cascade occurs in the brains of AD patients and contributes to the local inflammatory response triggered by senile plaque. The existence of an inflammatory component in AD is now well known on the basis of epidemiological findings showing a reduced prevalence of the disease upon long-term medication with anti-inflammatory drugs, and evidence from studies of clinical materials that shows an accumulation of activated glial cells, particularly microglia and astrocytes, in the same areas as amyloid plaques. Glial cells maintain brain plasticity and protect the brain for functional recovery from injuries. Dysfunction of glial cells may promote neurodegeneration and, eventually, the retraction of neuronal synapses, which leads to cognitive deficits. The focus of this review is on glial cells and their diversity properties in AD.
Figures
References
-
- Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–8. - PubMed
-
- Heinonen O, Soininen H, Sorvari H, Kosunen O, Paljärvi L, Koivisto E, Riekkinen PJ., Sr Loss of synaptophysin-like immunoreactivity in the hippocampal formation is an early phenomenon in Alzheimer's disease. Neuroscience. 1995;64:375–84. - PubMed
-
- Jellinger KA, Bancher C. AD neuropathology. Neurology. 1996;46:1186–7. - PubMed
-
- Goldman JS, Reed B, Gearhart R, Kramer JH, Miller BL. Very early-onset familial Alzheimer's disease: a novel presenilin 1 mutation. Int J Geriatr Psychiatry. 2002;17:649–51. - PubMed
-
- Wisniewski T, Ghiso J, Frangione B. Alzheimer's disease and soluble A beta. Neurobiol Aging. 1994;15:143–52. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
