

The deleted in brachydactyly B domain of ROR2 is required for receptor activation by recruitment of Src
- PMID: 18365018
- PMCID: PMC2268744
- DOI: 10.1371/journal.pone.0001873
The deleted in brachydactyly B domain of ROR2 is required for receptor activation by recruitment of Src
Abstract
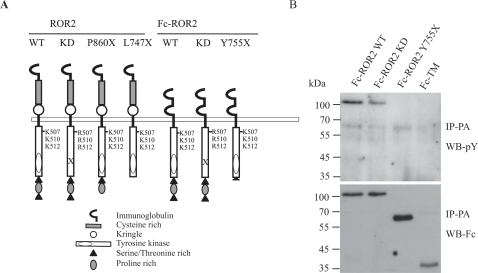
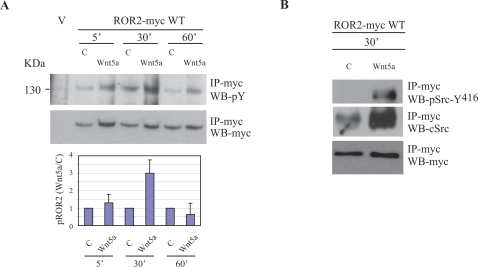
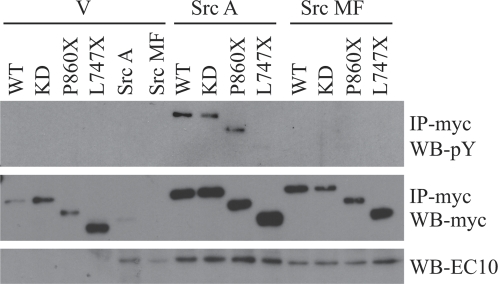
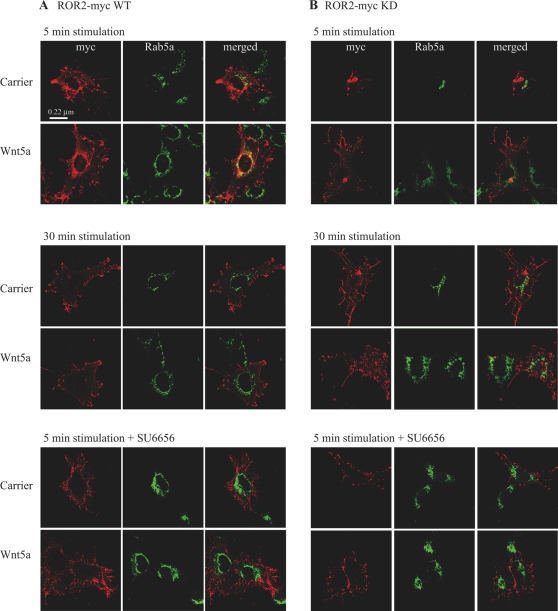
The transmembrane receptor 'ROR2' resembles members of the receptor tyrosine kinase family of signalling receptors in sequence but its' signal transduction mechanisms remain enigmatic. This problem has particular importance because mutations in ROR2 are associated with two human skeletal dysmorphology syndromes, recessive Robinow Syndrome (RS) and dominant acting Brachydactyly type B (BDB). Here we show, using a constitutive dimerisation approach, that ROR2 exhibits dimerisation-induced tyrosine kinase activity and the ROR2 C-terminal domain, which is deleted in BDB, is required for recruitment and activation of the non-receptor tyrosine kinase Src. Native ROR2 phosphorylation is induced by the ligand Wnt5a and is blocked by pharmacological inhibition of Src kinase activity. Eight sites of Src-mediated ROR2 phosphorylation have been identified by mass spectrometry. Activation via tyrosine phosphorylation of ROR2 receptor leads to its internalisation into Rab5 positive endosomes. These findings show that BDB mutant receptors are defective in kinase activation as a result of failure to recruit Src.
Conflict of interest statement
Figures

References
-
- Masiakowski P, Carroll RD. A novel family of cell surface receptors with tyrosine kinase-like domain. J Biol Chem. 1992;267:26181–26190. - PubMed
-
- Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat Genet. 2000;25:419–422. - PubMed
-
- Oldridge M, Fortuna AM, Maringa M, Propping P, Mansour S, et al. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nat Genet. 2000;24:275–278. - PubMed
-
- van Bokhoven H, Celli J, Kayserili H, van Beusekom E, Balci S, et al. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat Genet. 2000;25:423–426. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
