

Transcription network construction for large-scale microarray datasets using a high-performance computing approach
- PMID: 18366618
- PMCID: PMC2386070
- DOI: 10.1186/1471-2164-9-S1-S5
Transcription network construction for large-scale microarray datasets using a high-performance computing approach
Abstract
Background: The advance in high-throughput genomic technologies including microarrays has demonstrated the potential of generating a tremendous amount of gene expression data for the entire genome. Deciphering transcriptional networks that convey information on intracluster correlations and intercluster connections of genes is a crucial analysis task in the post-sequence era. Most of the existing analysis methods for genome-wide gene expression profiles consist of several steps that often require human involvement based on experiential knowledge that is generally difficult to acquire and formalize. Moreover, large-scale datasets typically incur prohibitively expensive computation overhead and thus result in a long experiment-analysis research cycle.
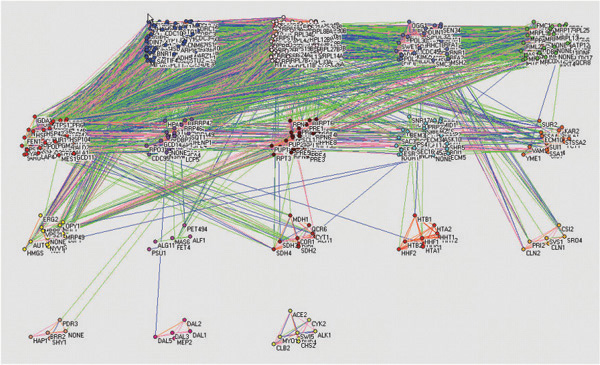
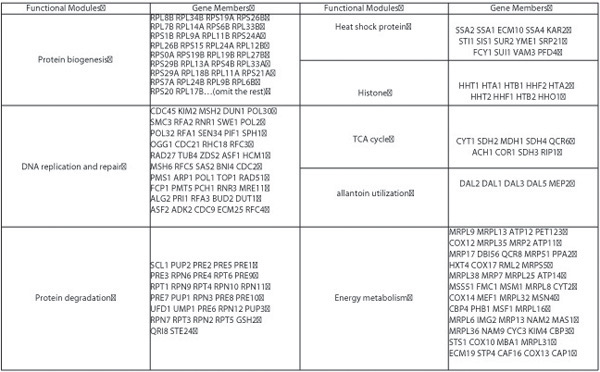
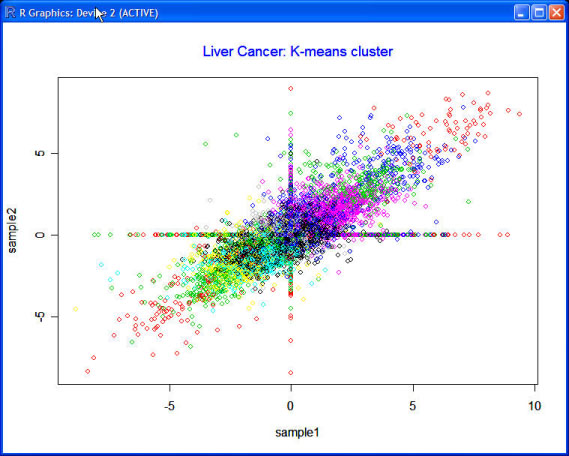
Results: We propose a parallel computation-based random matrix theory approach to analyze the cross correlations of gene expression data in an entirely automatic and objective manner to eliminate the ambiguities and subjectivity inherent to human decisions. We apply the proposed approach to the publicly available human liver cancer data and yeast cycle data, and generate transcriptional networks that illustrate interacting functional modules. The experimental results conform accurately to those published in previous literatures.
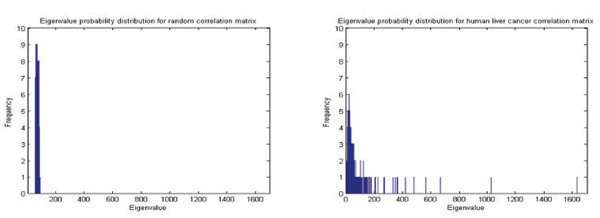
Conclusions: The correlations calculated from experimental measurements typically contain both "genuine" and "random" components. In the proposed approach, we remove the "random" component by testing the statistics of the eigenvalues of the correlation matrix against a "null hypothesis"--a truly random correlation matrix obtained from mutually uncorrelated expression data series. Our investigation into the components of deviating eigenvectors after varimax orthogonal rotation reveals distinct functional modules. The utilization of high performance computing resources including ScaLAPACK package, supercomputer and Linux PC cluster in our implementations and experiments significantly reduces the amount of computation time that is otherwise needed on a single workstation. More importantly, the large distributed shared memory and parallel computing power allow us to process genomic datasets of enormous sizes.
Figures
References
-
- Liang S, Fuhrman S, Somogyi R. REVEAL, a general reverse engineering algorithm for inference of genetic network architectures. Pacific Symp Biocomp. 1998;3:18–29. - PubMed
-
- Akutsu T, Miyano S, Kuhara S. Identification of genetic networks from a small number of gene expression patterns under theboolean network model. Pacific Symp Biocomp. 1986;4:17–28. - PubMed
-
- Chen T, He H, Church G. Modeling gene expression with differential equations. Pacific Symp Biocomp. 1999;4:29–40. - PubMed
-
- Sokal R, Michener C. A statistical method for evaluating systematic relationships. Univ Kansas Sci Bull. 1958;38:1409–1438.
-
- MacQueen JB. Some Methods for classification and Analysis of Multivariate Observations. Proceedings of 5-th Berkeley Symposium on Mathematical Statistics and Probability. 1998;1:281–297.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
