

Prediction of human disease genes by human-mouse conserved coexpression analysis
- PMID: 18369433
- PMCID: PMC2268251
- DOI: 10.1371/journal.pcbi.1000043
Prediction of human disease genes by human-mouse conserved coexpression analysis
Abstract
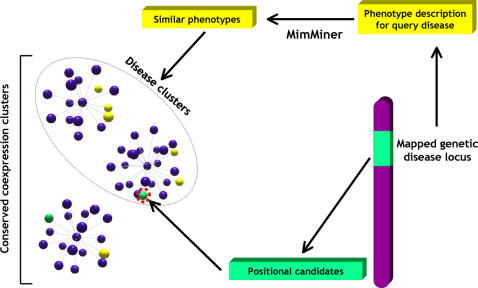
Background: Even in the post-genomic era, the identification of candidate genes within loci associated with human genetic diseases is a very demanding task, because the critical region may typically contain hundreds of positional candidates. Since genes implicated in similar phenotypes tend to share very similar expression profiles, high throughput gene expression data may represent a very important resource to identify the best candidates for sequencing. However, so far, gene coexpression has not been used very successfully to prioritize positional candidates.
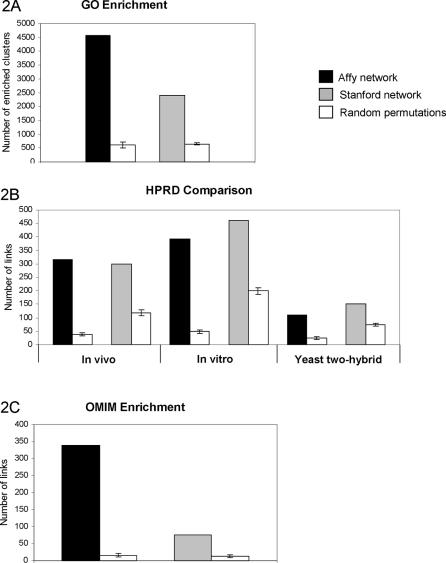
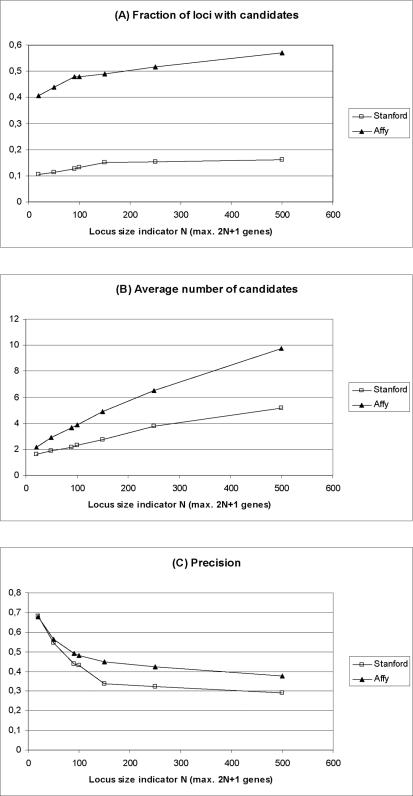
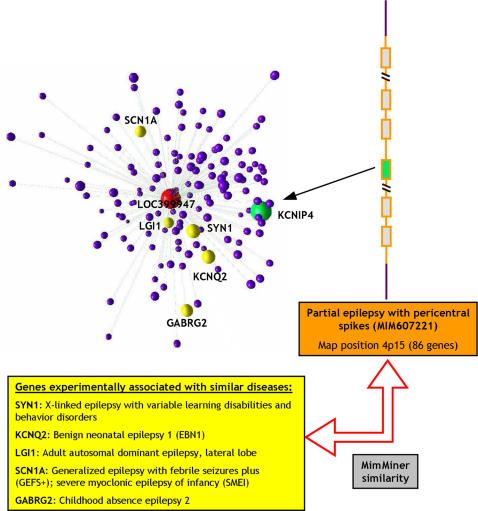
Methodology/principal findings: We show that it is possible to reliably identify disease-relevant relationships among genes from massive microarray datasets by concentrating only on genes sharing similar expression profiles in both human and mouse. Moreover, we show systematically that the integration of human-mouse conserved coexpression with a phenotype similarity map allows the efficient identification of disease genes in large genomic regions. Finally, using this approach on 850 OMIM loci characterized by an unknown molecular basis, we propose high-probability candidates for 81 genetic diseases.
Conclusion: Our results demonstrate that conserved coexpression, even at the human-mouse phylogenetic distance, represents a very strong criterion to predict disease-relevant relationships among human genes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
