

A generic approach to identify Transcription Factor-specific operator motifs; Inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1
- PMID: 18371204
- PMCID: PMC2329647
- DOI: 10.1186/1471-2164-9-145
A generic approach to identify Transcription Factor-specific operator motifs; Inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1
Abstract
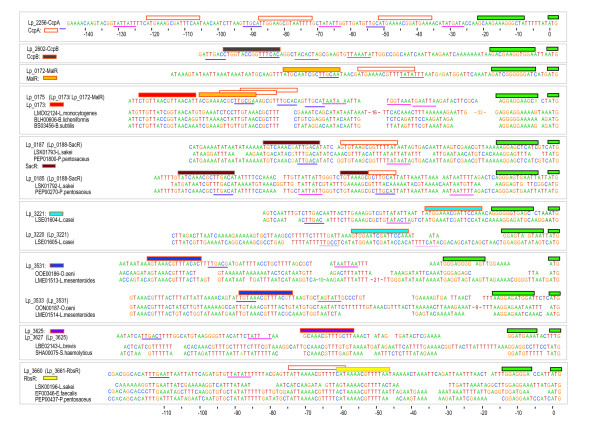
Background: A key problem in the sequence-based reconstruction of regulatory networks in bacteria is the lack of specificity in operator predictions. The problem is especially prominent in the identification of transcription factor (TF) specific binding sites. More in particular, homologous TFs are abundant and, as they are structurally very similar, it proves difficult to distinguish the related operators by automated means. This also holds for the LacI-family, a family of TFs that is well-studied and has many members that fulfill crucial roles in the control of carbohydrate catabolism in bacteria including catabolite repression. To overcome the specificity problem, a comprehensive footprinting approach was formulated to identify TF-specific operator motifs and was applied to the LacI-family of TFs in the model gram positive organism, Lactobacillus plantarum WCFS1. The main premise behind the approach is that only orthologous sequences that share orthologous genomic context will share equivalent regulatory sites.
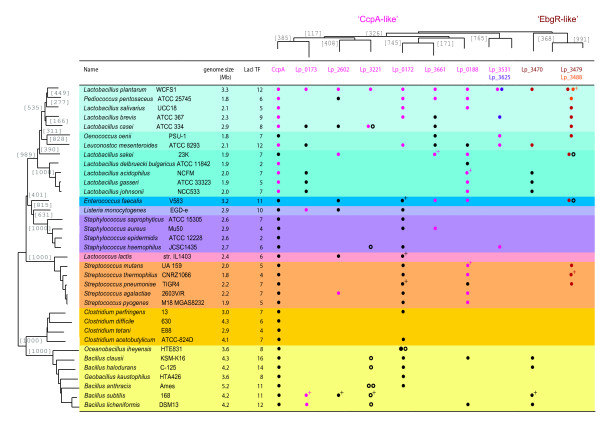
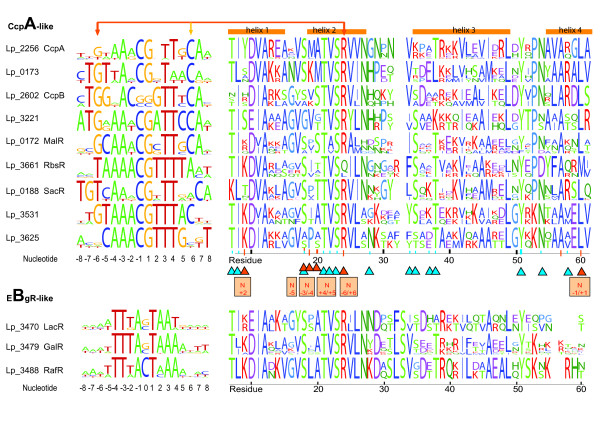
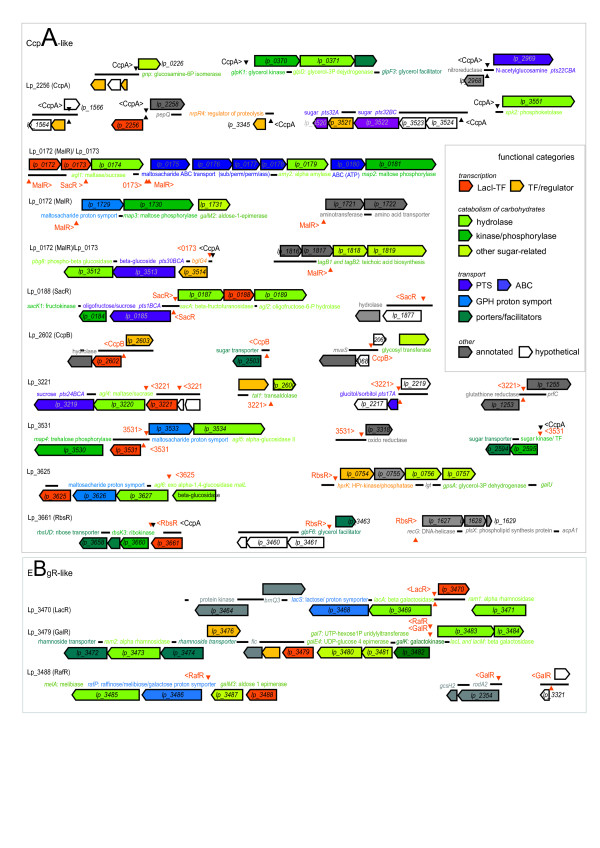
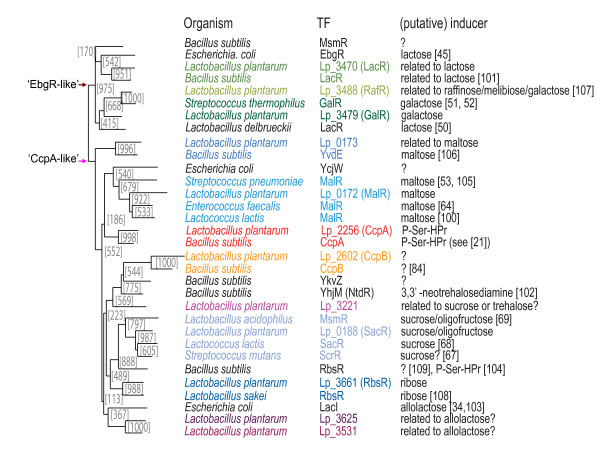
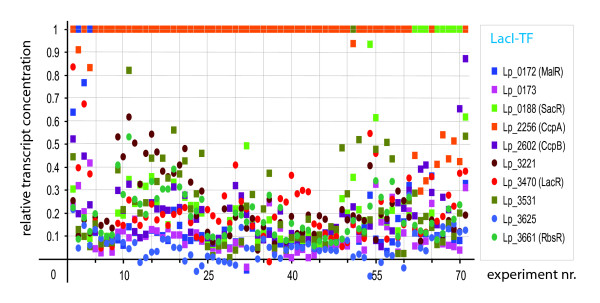
Results: When the approach was applied to the 12 LacI-family TFs of the model species, a specific operator motif was identified for each of them. With the TF-specific operator motifs, potential binding sites were found on the genome and putative minimal regulons could be defined. Moreover, specific inducers could in most cases be linked to the TFs through phylogeny, thereby unveiling the biological role of these regulons. The operator predictions indicated that the LacI-family TFs can be separated into two subfamilies with clearly distinct operator motifs. They also established that the operator related to the 'global' regulator CcpA is not inherently distinct from that of other LacI-family members, only more degenerate. Analysis of the chromosomal position of the identified putative binding sites confirmed that the LacI-family TFs are mostly auto-regulatory and relate mainly to carbohydrate uptake and catabolism.
Conclusion: Our approach to identify specific operator motifs for different TF-family members is specific and in essence generic. The data infer that, although the specific operator motifs can be used to identify minimal regulons, experimental knowledge on TF activity especially is essential to determine complete regulons as well as to estimate the overlap between TF affinities.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
