

Hematopoietic fingerprints: an expression database of stem cells and their progeny
- PMID: 18371395
- PMCID: PMC2475548
- DOI: 10.1016/j.stem.2007.10.003
Hematopoietic fingerprints: an expression database of stem cells and their progeny
Abstract
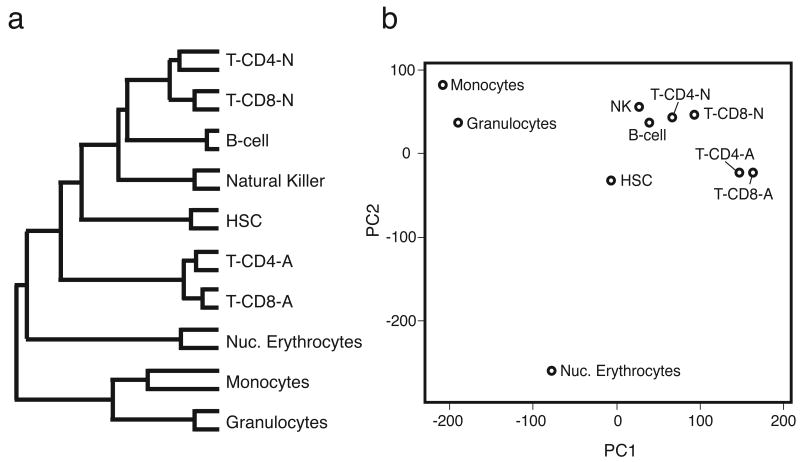
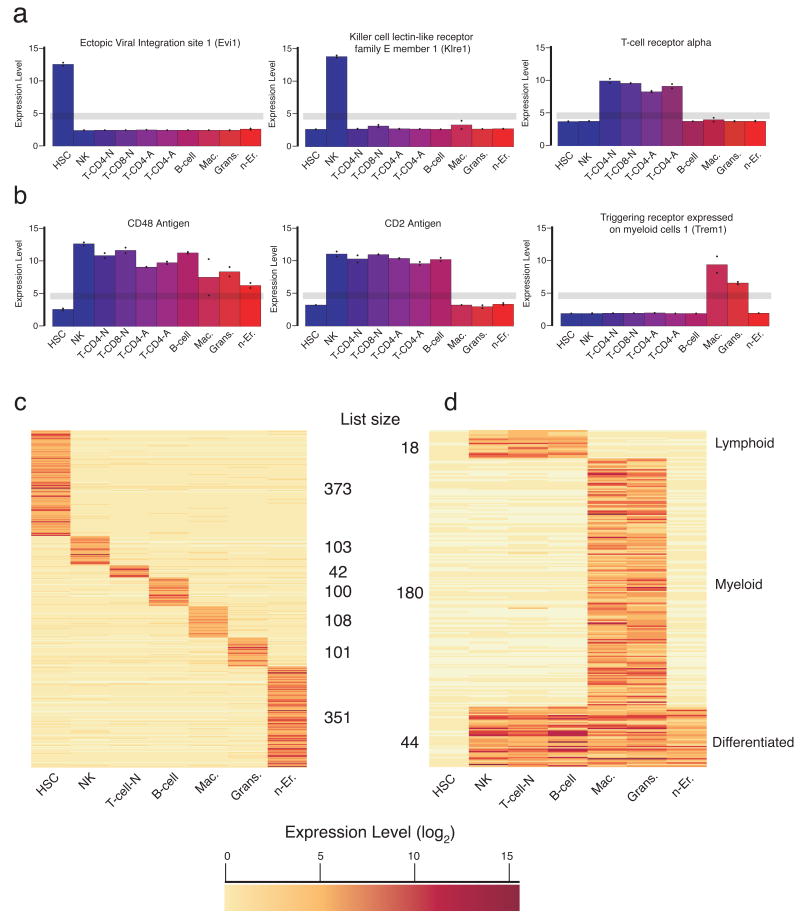
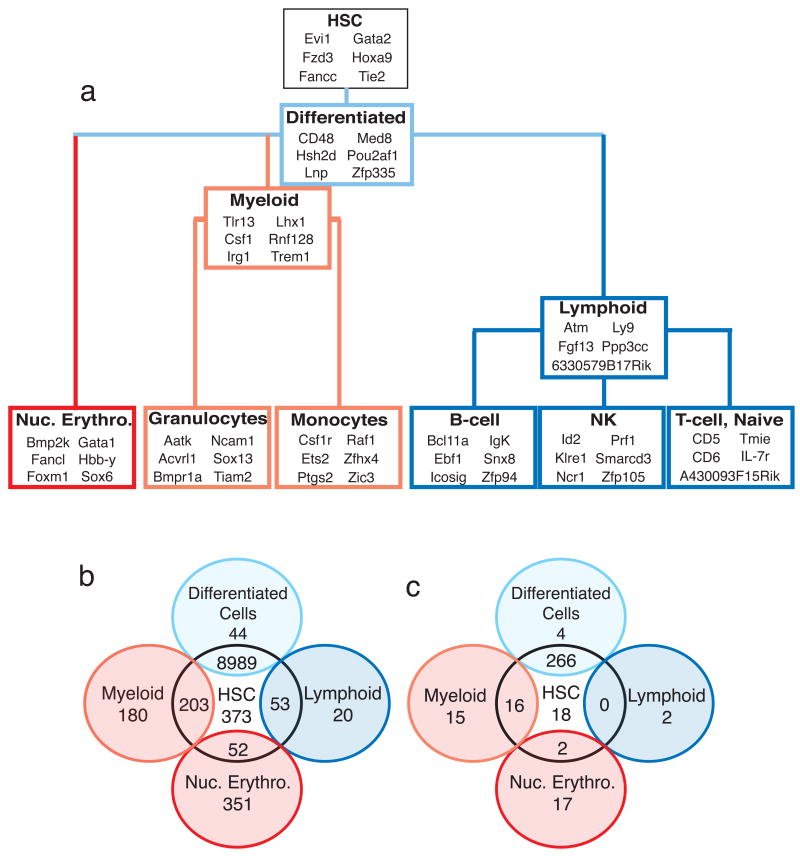
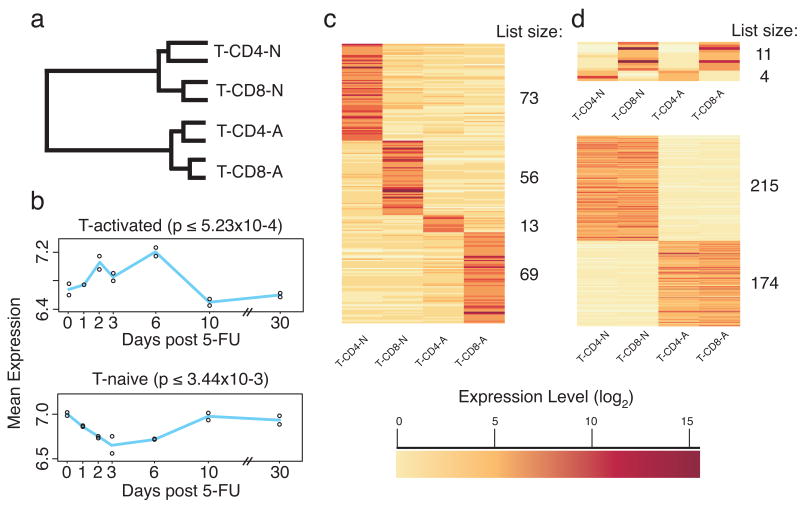
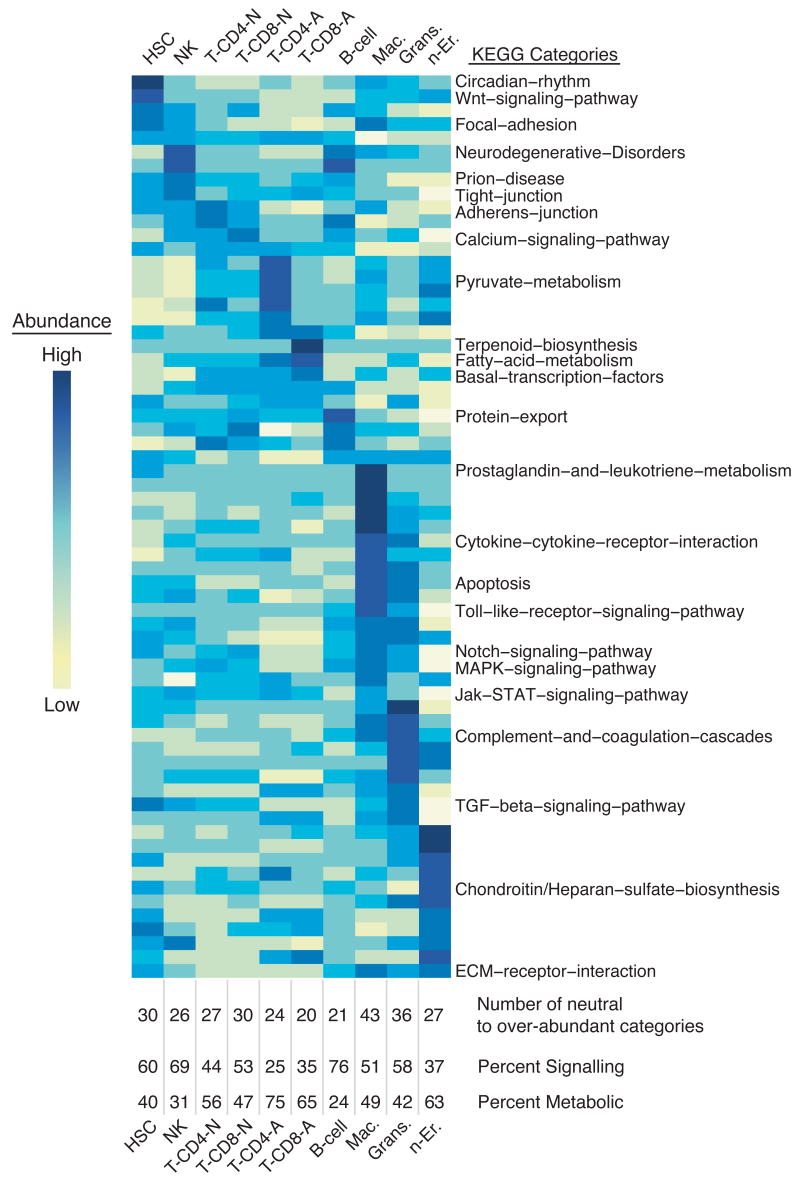
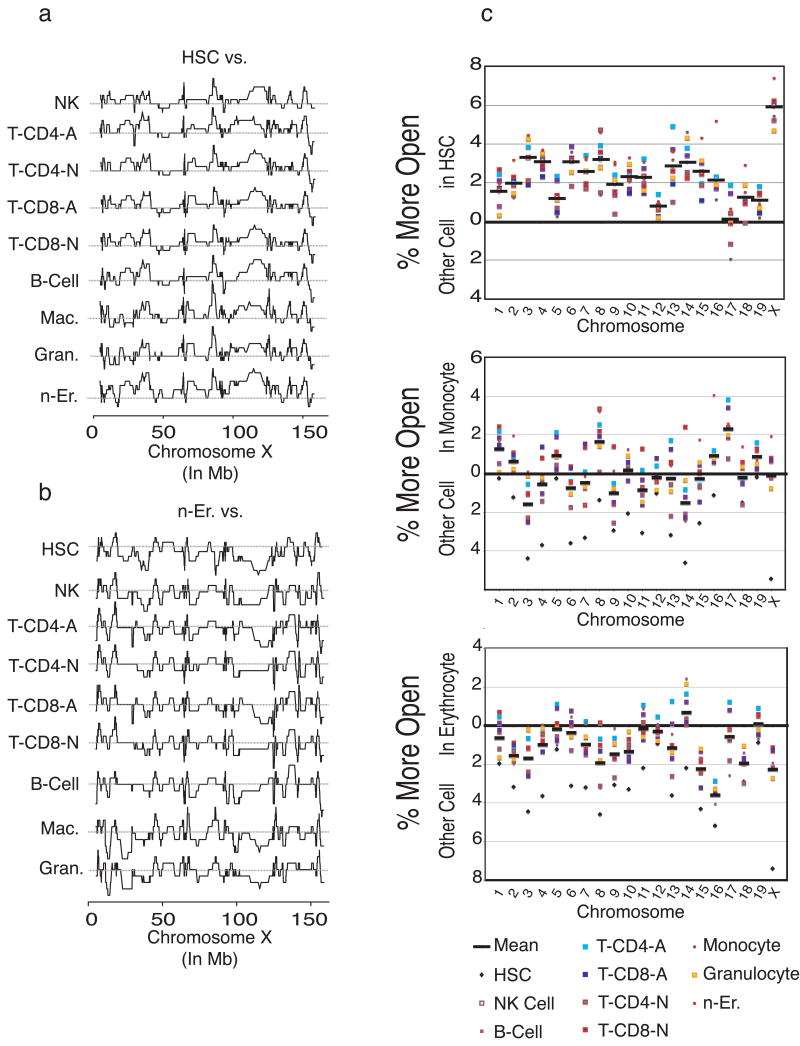
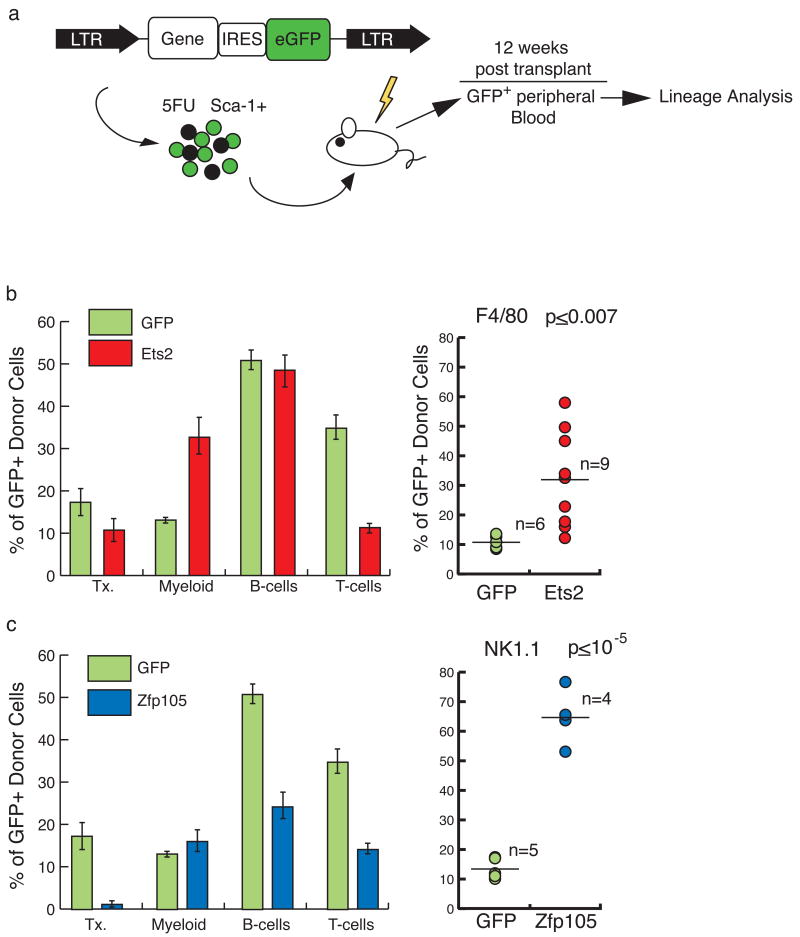
Hematopoietic stem cells (HSCs) continuously regenerate the hematologic system, yet few genes regulating this process have been defined. To identify candidate factors involved in differentiation and self-renewal, we have generated an expression database of hematopoietic stem cells and their differentiated progeny, including erythrocytes, granulocytes, monocytes, NK cells, activated and naive T cells, and B cells. Bioinformatic analysis revealed HSCs were more transcriptionally active than their progeny and shared a common activation mechanism with T cells. Each cell type also displayed unique biases in the regulation of particular genetic pathways, with Wnt signaling particularly enhanced in HSCs. We identified approximately 100-400 genes uniquely expressed in each cell type, termed lineage "fingerprints." In overexpression studies, two of these genes, Zfp 105 from the NK cell lineage, and Ets2 from the monocyte lineage, were able to significantly influence differentiation toward their respective lineages, demonstrating the utility of the fingerprints for identifying genes that regulate differentiation.
Figures
References
-
- Adolfsson J, Mansson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, Bryder D, Yang L, Borge OJ, Thoren LA, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121:295–306. - PubMed
-
- Akashi K, He X, Chen J, Iwasaki H, Niu C, Steenhard B, Zhang J, Haug J, Li L. Transcriptional accessibility for genes of multiple tissues and hematopoietic lineages is hierarchically controlled during early hematopoiesis. Blood. 2003;101:383–389. - PubMed
-
- Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. - PubMed
-
- Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
