

SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization
- PMID: 18371449
- PMCID: PMC2819008
- DOI: 10.1016/j.stem.2008.01.002
SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization
Abstract
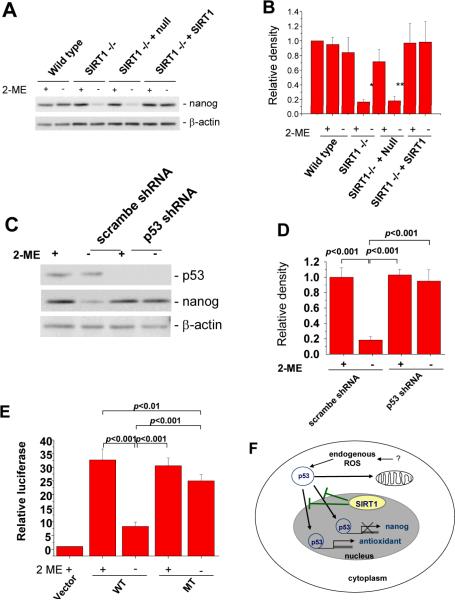
Nuclear tumor suppressor p53 transactivates proapoptotic genes or antioxidant genes depending on stress severity, while cytoplasmic p53 induces mitochondrial-dependent apoptosis without gene transactivation. Although SIRT1, a p53 deacetylase, inhibits p53-mediated transactivation, how SIRT1 regulates these p53 multifunctions is unclear. Here we show that SIRT1 blocks nuclear translocation of cytoplasmic p53 in response to endogenous reactive oxygen species (ROS) and triggers mitochondrial-dependent apoptosis in mouse embryonic stem (mES) cells. ROS generated by antioxidant-free culture caused p53 translocation into mitochondria in wild-type mES cells but induced p53 translocation into the nucleus in SIRT1(-/-) mES cells. Endogenous ROS triggered apoptosis of wild-type mES through mitochondrial translocation of p53 and BAX but inhibited Nanog expression of SIRT1(-/-) mES, indicating that SIRT1 makes mES cells sensitive to ROS and inhibits p53-mediated suppression of Nanog expression. Our results suggest that endogenous ROS control is important for mES cell maintenance in culture.
Figures




References
-
- Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R, Wahl GM. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr. Biol. 1998;8:145–55. - PubMed
-
- Arai M, Yoguchi A, Takizawa T, Yokoyama T, Kanda T, Kurabayashi M, Nagai R. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca2+-ATPase gene transcription. Circ. Res. 2000;86:8–14. - PubMed
-
- Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A, Del Sal G, Syed N, Smith P, Gasco M, Crook T, Lu X. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat. Genet. 2006;38:1133–41. - PubMed
-
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat. Rev. Mol. Cell. Biol. 2005;6:298–305. - PubMed
-
- Braithwaite AW, Del Sal G, Lu X. Some p53-binding proteins that can function as arbiters of life and death. Cell Death Differ. 2006;13:984–93. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
