

Walking the interactome for prioritization of candidate disease genes
- PMID: 18371930
- PMCID: PMC2427257
- DOI: 10.1016/j.ajhg.2008.02.013
Walking the interactome for prioritization of candidate disease genes
Abstract
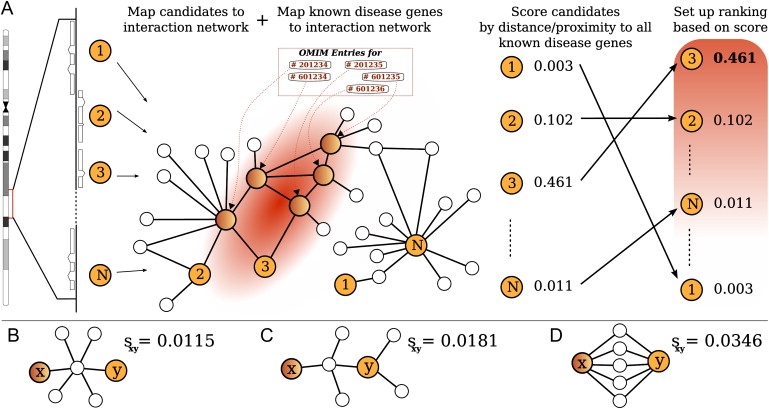
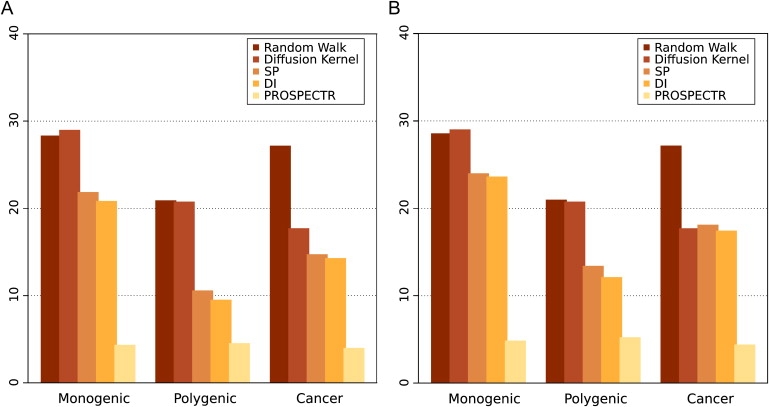
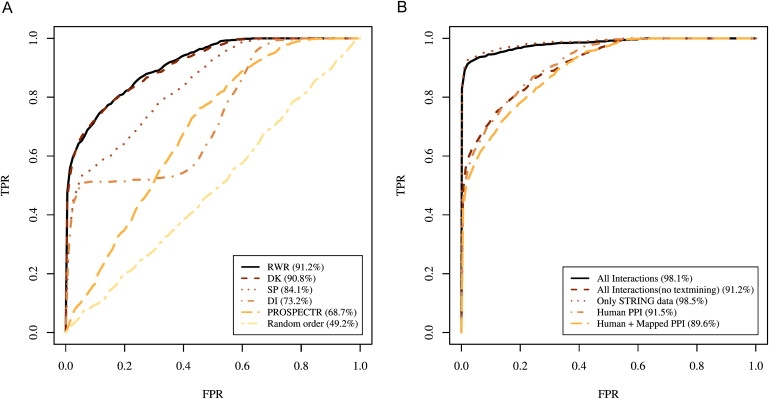
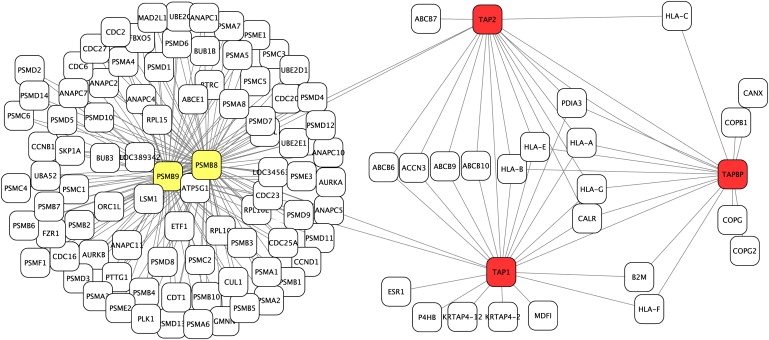
The identification of genes associated with hereditary disorders has contributed to improving medical care and to a better understanding of gene functions, interactions, and pathways. However, there are well over 1500 Mendelian disorders whose molecular basis remains unknown. At present, methods such as linkage analysis can identify the chromosomal region in which unknown disease genes are located, but the regions could contain up to hundreds of candidate genes. In this work, we present a method for prioritization of candidate genes by use of a global network distance measure, random walk analysis, for definition of similarities in protein-protein interaction networks. We tested our method on 110 disease-gene families with a total of 783 genes and achieved an area under the ROC curve of up to 98% on simulated linkage intervals of 100 genes surrounding the disease gene, significantly outperforming previous methods based on local distance measures. Our results not only provide an improved tool for positional-cloning projects but also add weight to the assumption that phenotypically similar diseases are associated with disturbances of subnetworks within the larger protein interactome that extend beyond the disease proteins themselves.
Figures
References
-
- Brunner H.G., van Driel M.A. From syndrome families to functional genomics. Nat. Rev. Genet. 2004;5:545–551. - PubMed
-
- Glazier A.M., Nadeau J.H., Aitman T.J. Finding genes that underlie complex traits. Science. 2002;298:2345–2349. - PubMed
-
- Botstein D., Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003;33(Suppl):228–237. - PubMed
-
- Perez-Iratxeta C., Bork P., Andrade M.A. Association of genes to genetically inherited diseases using data mining. Nat. Genet. 2002;31:316–319. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
