

Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia
- PMID: 18371931
- PMCID: PMC2427297
- DOI: 10.1016/j.ajhg.2008.02.017
Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia
Abstract
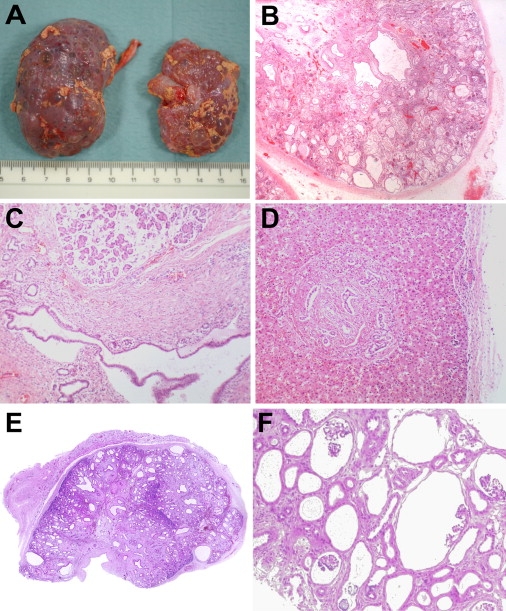
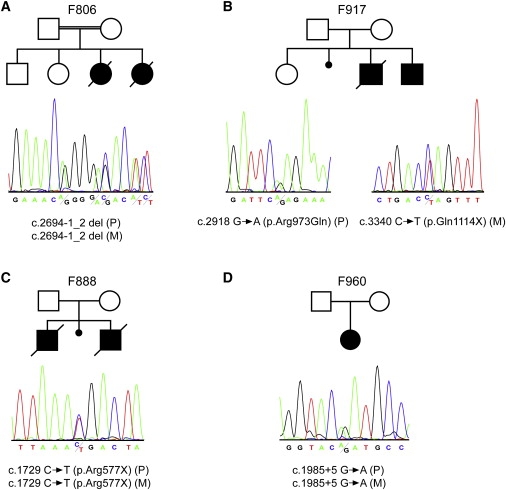
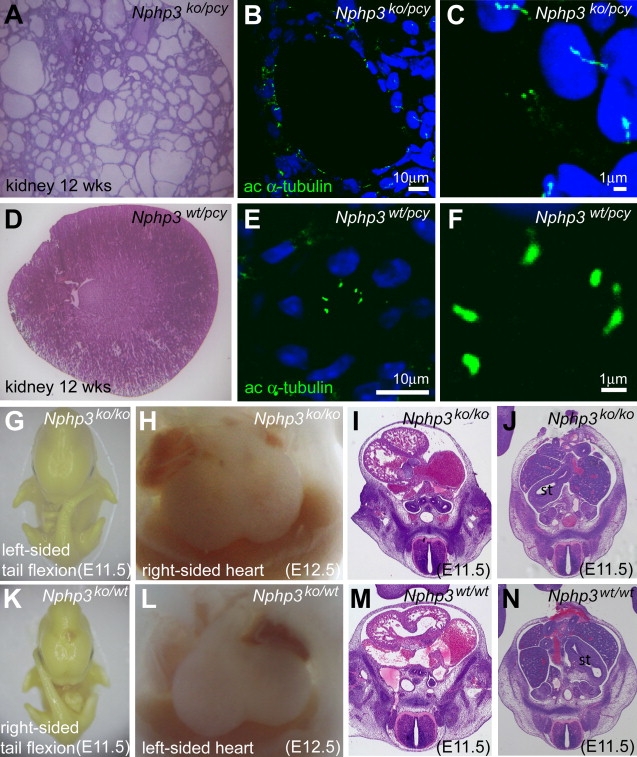
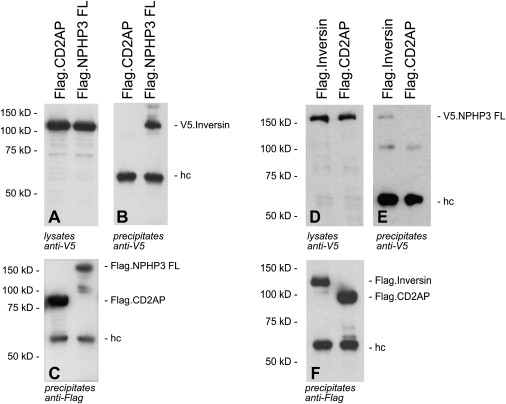
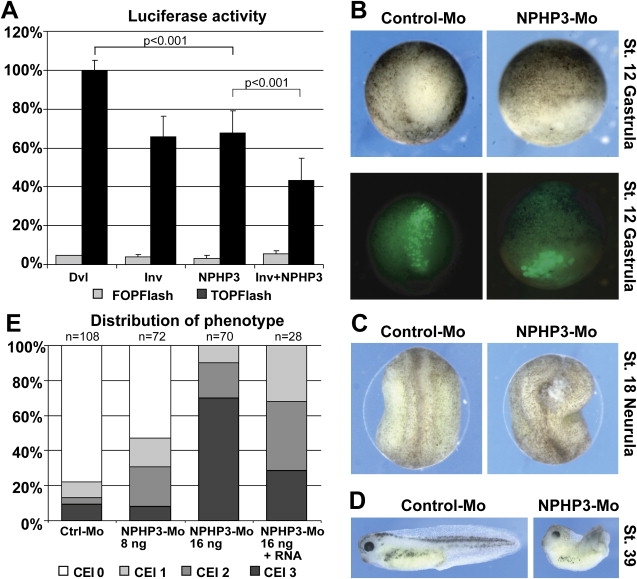
Many genetic diseases have been linked to the dysfunction of primary cilia, which occur nearly ubiquitously in the body and act as solitary cellular mechanosensory organelles. The list of clinical manifestations and affected tissues in cilia-related disorders (ciliopathies) such as nephronophthisis is broad and has been attributed to the wide expression pattern of ciliary proteins. However, little is known about the molecular mechanisms leading to this dramatic diversity of phenotypes. We recently reported hypomorphic NPHP3 mutations in children and young adults with isolated nephronophthisis and associated hepatic fibrosis or tapetoretinal degeneration. Here, we chose a combinatorial approach in mice and humans to define the phenotypic spectrum of NPHP3/Nphp3 mutations and the role of the nephrocystin-3 protein. We demonstrate that the pcy mutation generates a hypomorphic Nphp3 allele that is responsible for the cystic kidney disease phenotype, whereas complete loss of Nphp3 function results in situs inversus, congenital heart defects, and embryonic lethality in mice. In humans, we show that NPHP3 mutations can cause a broad clinical spectrum of early embryonic patterning defects comprising situs inversus, polydactyly, central nervous system malformations, structural heart defects, preauricular fistulas, and a wide range of congenital anomalies of the kidney and urinary tract (CAKUT). On the functional level, we show that nephrocystin-3 directly interacts with inversin and can inhibit like inversin canonical Wnt signaling, whereas nephrocystin-3 deficiency leads in Xenopus laevis to typical planar cell polarity defects, suggesting a role in the control of canonical and noncanonical (planar cell polarity) Wnt signaling.
Figures
Similar articles
-
Nephrocystin-3 is required for ciliary function in zebrafish embryos.Am J Physiol Renal Physiol. 2010 Jul;299(1):F55-62. doi: 10.1152/ajprenal.00043.2010. Epub 2010 May 12. Am J Physiol Renal Physiol. 2010. PMID: 20462968 Free PMC article.
-
Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination.Nat Genet. 2003 Aug;34(4):413-20. doi: 10.1038/ng1217. Nat Genet. 2003. PMID: 12872123 Free PMC article.
-
Caroli disease, bilateral diffuse cystic renal dysplasia, situs inversus, postaxial polydactyly, and preauricular fistulas: a ciliopathy caused by a homozygous NPHP3 mutation.Eur J Pediatr. 2013 Jul;172(7):877-81. doi: 10.1007/s00431-011-1552-0. Epub 2011 Aug 16. Eur J Pediatr. 2013. PMID: 21845392
-
Nephronophthisis: disease mechanisms of a ciliopathy.J Am Soc Nephrol. 2009 Jan;20(1):23-35. doi: 10.1681/ASN.2008050456. Epub 2008 Dec 31. J Am Soc Nephrol. 2009. PMID: 19118152 Free PMC article. Review.
-
Renal-hepatic-pancreatic dysplasia-1 with a novel NPHP3 genotype: a case report and review of the literature.BMC Pediatr. 2022 Oct 18;22(1):603. doi: 10.1186/s12887-022-03659-7. BMC Pediatr. 2022. PMID: 36253741 Free PMC article. Review.
Cited by
-
Renal cell carcinoma in an adult-onset ESRD patient with nephronophthisis harboring NPHP3 deletion: A case report.Heliyon. 2024 Mar 29;10(7):e28985. doi: 10.1016/j.heliyon.2024.e28985. eCollection 2024 Apr 15. Heliyon. 2024. PMID: 38617907 Free PMC article.
-
A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies.Nat Genet. 2009 Jun;41(6):739-45. doi: 10.1038/ng.366. Epub 2009 May 10. Nat Genet. 2009. PMID: 19430481 Free PMC article.
-
Cystic kidney disease: the role of Wnt signaling.Trends Mol Med. 2010 Aug;16(8):349-60. doi: 10.1016/j.molmed.2010.05.004. Epub 2010 Jun 22. Trends Mol Med. 2010. PMID: 20576469 Free PMC article. Review.
-
A meckelin-filamin A interaction mediates ciliogenesis.Hum Mol Genet. 2012 Mar 15;21(6):1272-86. doi: 10.1093/hmg/ddr557. Epub 2011 Nov 25. Hum Mol Genet. 2012. PMID: 22121117 Free PMC article.
-
The ciliopathies: a transitional model into systems biology of human genetic disease.Curr Opin Genet Dev. 2012 Jun;22(3):290-303. doi: 10.1016/j.gde.2012.04.006. Epub 2012 May 23. Curr Opin Genet Dev. 2012. PMID: 22632799 Free PMC article. Review.
References
-
- Hildebrandt F., Otto E. Cilia and centrosomes: A unifying pathogenic concept for cystic kidney disease? Nat. Rev. Genet. 2005;6:928–940. - PubMed
-
- Harris P.C., Torres V.E. Understanding pathogenic mechanisms in polycystic kidney disease provides clues for therapy. Curr. Opin. Nephrol. Hypertens. 2006;15:456–463. - PubMed
-
- Badano J.L., Mitsuma N., Beales P.L., Katsanis N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006;7:125–148. - PubMed
-
- Mykytyn K. Clinical variability in ciliary disorders. Nat. Genet. 2007;39:818–819. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
