

Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes
- PMID: 18372317
- PMCID: PMC2900904
- DOI: 10.1093/hmg/ddn099
Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes
Abstract
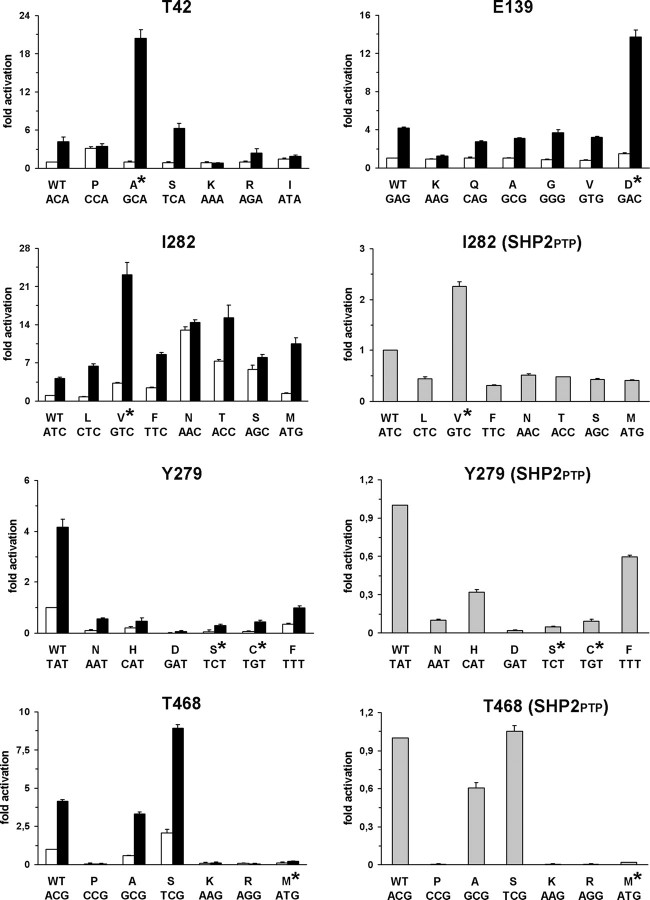
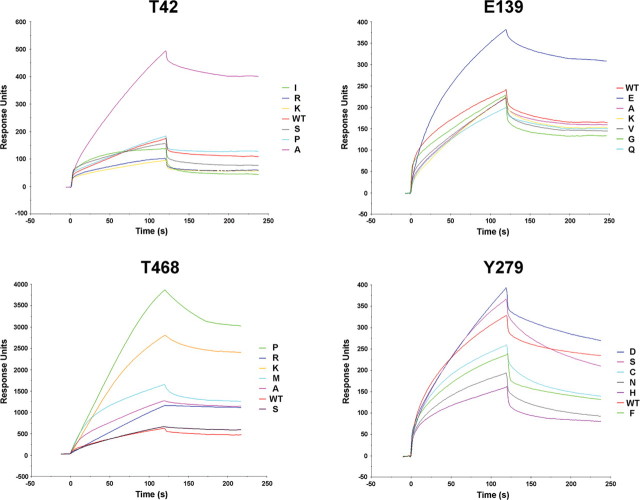
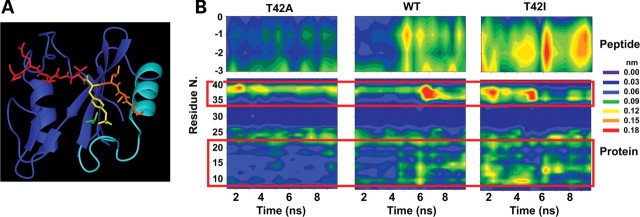
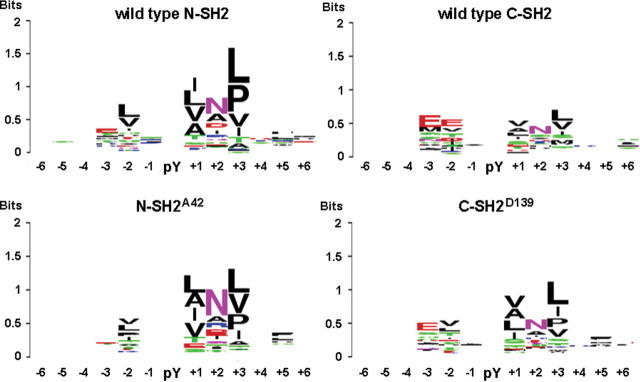
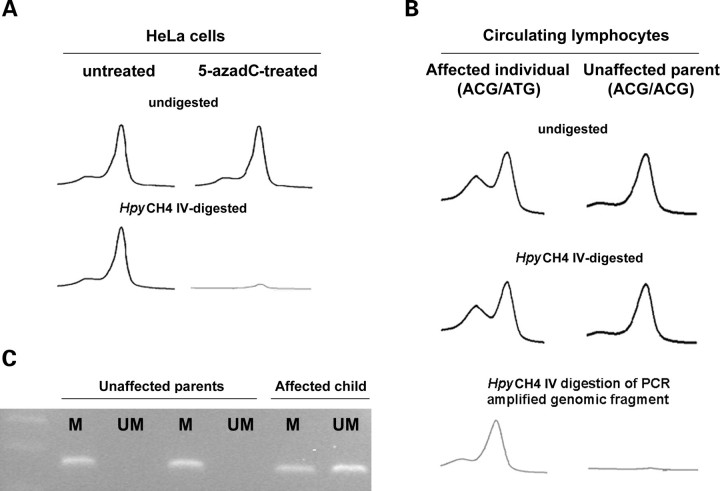
Missense PTPN11 mutations cause Noonan and LEOPARD syndromes (NS and LS), two developmental disorders with pleiomorphic phenotypes. PTPN11 encodes SHP2, an SH2 domain-containing protein tyrosine phosphatase functioning as a signal transducer. Generally, different substitutions of a particular amino acid residue are observed in these diseases, indicating that the crucial factor is the residue being replaced. For a few codons, only one substitution is observed, suggesting the possibility of specific roles for the residue introduced. We analyzed the biochemical behavior and ligand-binding properties of all possible substitutions arising from single-base changes affecting codons 42, 139, 279, 282 and 468 to investigate the mechanisms underlying the invariant occurrence of the T42A, E139D and I282V substitutions in NS and the Y279C and T468M changes in LS. Our data demonstrate that the isoleucine-to-valine change at codon 282 is the only substitution at that position perturbing the stability of SHP2's closed conformation without impairing catalysis, while the threonine-to-alanine change at codon 42, but not other substitutions of that residue, promotes increased phosphopeptide-binding affinity. The recognition specificity of the C-SH2 domain bearing the E139D substitution differed substantially from its wild-type counterpart acquiring binding properties similar to those observed for the N-SH2 domain, revealing a novel mechanism of SHP2's functional dysregulation. Finally, while functional selection does not seem to occur for the substitutions at codons 279 and 468, we point to deamination of the methylated cytosine at nucleotide 1403 as the driving factor leading to the high prevalence of the T468M change in LS.
Figures
References
-
- Tartaglia M., Mehler E.L., Goldberg R., Zampino G., Brunner H.G., Kremer H., van der Burgt I., Crosby A.H., Ion A., Jeffery S., et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP2, cause Noonan syndrome. Nat. Genet. 2001;29:465–468. - PubMed
-
- Noonan J. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am. J. Dis. Child. 1968;116:373–380. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
