

Cystic fibrosis transmembrane conductance regulator controls lung proteasomal degradation and nuclear factor-kappaB activity in conditions of oxidative stress
- PMID: 18372427
- PMCID: PMC2329829
- DOI: 10.2353/ajpath.2008.070310
Cystic fibrosis transmembrane conductance regulator controls lung proteasomal degradation and nuclear factor-kappaB activity in conditions of oxidative stress
Abstract
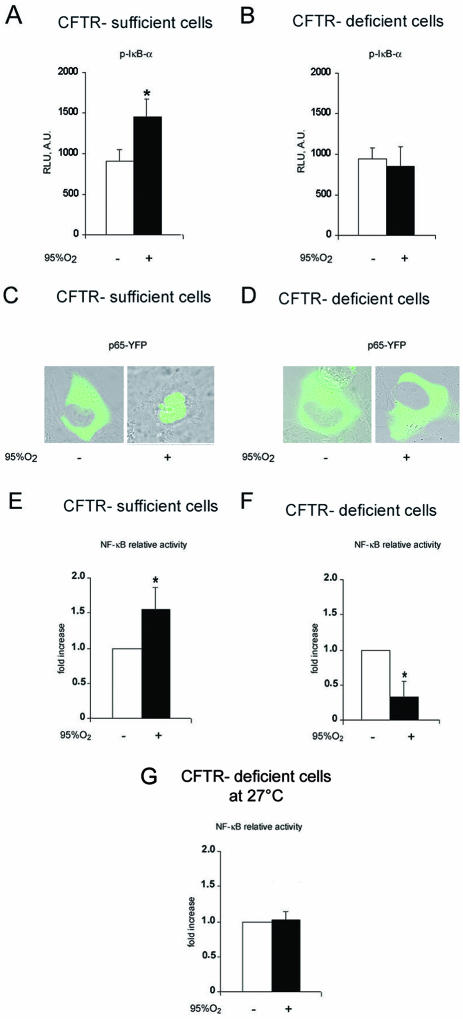
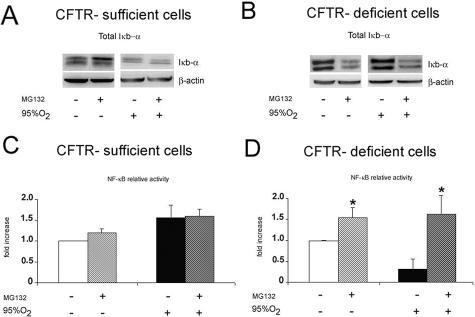
Cystic fibrosis is a lethal inherited disorder caused by mutations in a single gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein, resulting in progressive oxidative lung damage. In this study, we evaluated the role of CFTR in the control of ubiquitin-proteasome activity and nuclear factor (NF)-kappaB/IkappaB-alpha signaling after lung oxidative stress. After a 64-hour exposure to hyperoxia-mediated oxidative stress, CFTR-deficient (cftr(-/-)) mice exhibited significantly elevated lung proteasomal activity compared with wild-type (cftr(+/+)) animals. This was accompanied by reduced lung caspase-3 activity and defective degradation of NF-kappaB inhibitor IkappaB-alpha. In vitro, human CFTR-deficient lung cells exposed to oxidative stress exhibited increased proteasomal activity and decreased NF-kappaB-dependent transcriptional activity compared with CFTR-sufficient lung cells. Inhibition of the CFTR Cl(-) channel by CFTR(inh-172) in the normal bronchial immortalized cell line 16HBE14o- increased proteasomal degradation after exposure to oxidative stress. Caspase-3 inhibition by Z-DQMD in CFTR-sufficient lung cells mimicked the response profile of increased proteasomal degradation and reduced NF-kappaB activity observed in CFTR-deficient lung cells exposed to oxidative stress. Taken together, these results suggest that functional CFTR Cl(-) channel activity is crucial for regulation of lung proteasomal degradation and NF-kappaB activity in conditions of oxidative stress.
Figures




Similar articles
-
Resistance to Pseudomonas aeruginosa chronic lung infection requires cystic fibrosis transmembrane conductance regulator-modulated interleukin-1 (IL-1) release and signaling through the IL-1 receptor.Infect Immun. 2007 Apr;75(4):1598-608. doi: 10.1128/IAI.01980-06. Epub 2007 Feb 5. Infect Immun. 2007. PMID: 17283089 Free PMC article.
-
Parthenolide inhibits IkappaB kinase, NF-kappaB activation, and inflammatory response in cystic fibrosis cells and mice.Am J Respir Cell Mol Biol. 2007 Jun;36(6):728-36. doi: 10.1165/rcmb.2006-0323OC. Epub 2007 Feb 1. Am J Respir Cell Mol Biol. 2007. PMID: 17272824 Free PMC article.
-
Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice.Am J Respir Cell Mol Biol. 2006 Nov;35(5):579-86. doi: 10.1165/rcmb.2005-0473OC. Epub 2006 Jun 8. Am J Respir Cell Mol Biol. 2006. PMID: 16763223 Free PMC article.
-
Rethinking cystic fibrosis pathology: the critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation.Free Radic Biol Med. 2001 Jun 15;30(12):1440-61. doi: 10.1016/s0891-5849(01)00530-5. Free Radic Biol Med. 2001. PMID: 11390189 Review.
-
Defects in processing and trafficking of cystic fibrosis transmembrane conductance regulator.Exp Nephrol. 2000 Nov-Dec;8(6):332-42. doi: 10.1159/000020687. Exp Nephrol. 2000. PMID: 11014930 Review.
Cited by
-
CFTR Regulates the Proliferation, Migration and Invasion of Cervical Cancer Cells by Inhibiting the NF-κB Signalling Pathway.Cancer Manag Res. 2020 Jun 18;12:4685-4697. doi: 10.2147/CMAR.S252296. eCollection 2020. Cancer Manag Res. 2020. PMID: 32606960 Free PMC article.
-
Cystic Fibrosis: Systems Biology Analysis from Homozygous p.Phe508del Variant Patients' Samples Reveals Perturbations in Tissue-Specific Pathways.Biomed Res Int. 2021 Dec 2;2021:5262000. doi: 10.1155/2021/5262000. eCollection 2021. Biomed Res Int. 2021. PMID: 34901273 Free PMC article.
-
Loss of CFTR results in reduction of histone deacetylase 2 in airway epithelial cells.Am J Physiol Lung Cell Mol Physiol. 2009 Jul;297(1):L35-43. doi: 10.1152/ajplung.90399.2008. Epub 2009 May 1. Am J Physiol Lung Cell Mol Physiol. 2009. PMID: 19411311 Free PMC article.
-
The COPD-Associated Polymorphism Impairs the CFTR Function to Suppress Excessive IL-8 Production upon Environmental Pathogen Exposure.Int J Mol Sci. 2023 Jan 24;24(3):2305. doi: 10.3390/ijms24032305. Int J Mol Sci. 2023. PMID: 36768629 Free PMC article.
-
Oxidative stress, autophagy and airway ion transport.Am J Physiol Cell Physiol. 2019 Jan 1;316(1):C16-C32. doi: 10.1152/ajpcell.00341.2018. Epub 2018 Oct 10. Am J Physiol Cell Physiol. 2019. PMID: 30303690 Free PMC article. Review.
References
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA [published erratum appears in Science 1989 Sep 29;245(4925):1437]. Science. 1989;245:1066–1073. - PubMed
-
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. - PubMed
-
- Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. - PubMed
-
- Brown RK, McBurney A, Lunec J, Kelly FJ. Oxidative damage to DNA in patients with cystic fibrosis. Free Radic Biol Med. 1995;18:801–806. - PubMed
-
- Witko-Sarsat V, Allen RC, Paulais M, Nguyen AT, Bessou G, Lenoir G, Descamps-Latscha B. Disturbed myeloperoxidase-dependent activity of neutrophils in cystic fibrosis homozygotes and heterozygotes, and its correction by amiloride. J Immunol. 1996;157:2728–2735. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
