

Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy
- PMID: 18378900
- PMCID: PMC2448817
- DOI: 10.1073/pnas.0708100105
Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy
Abstract
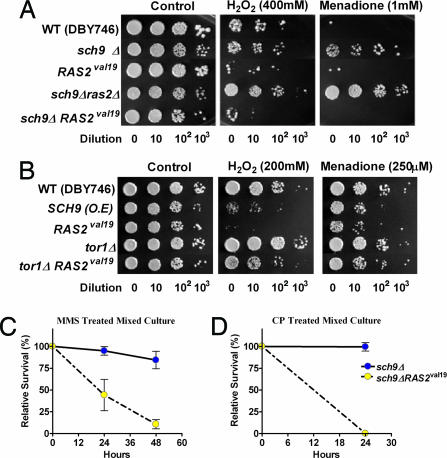
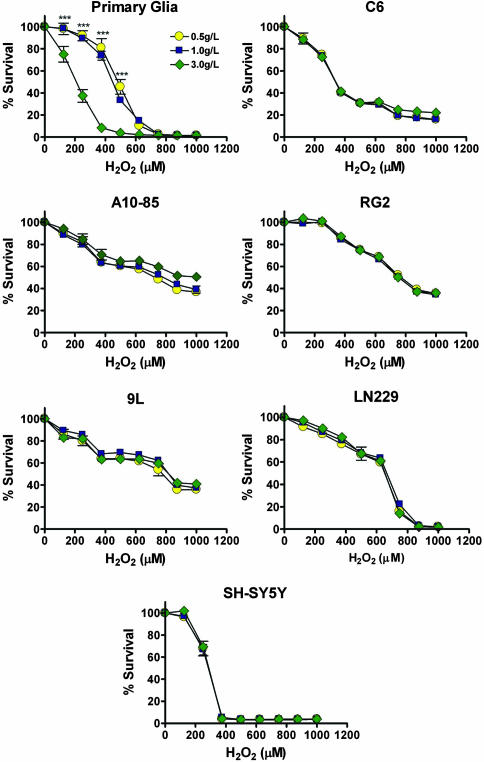
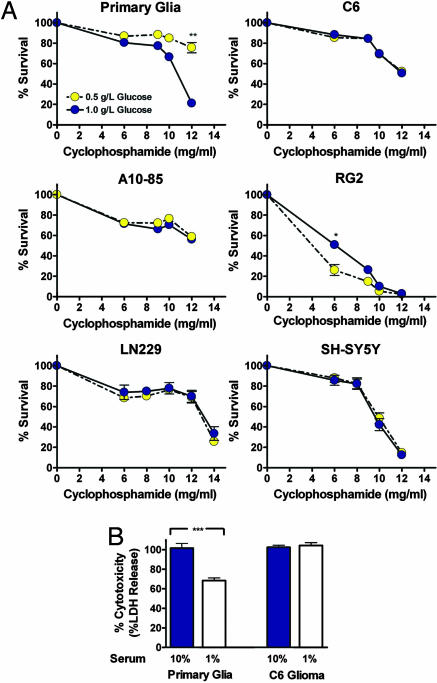
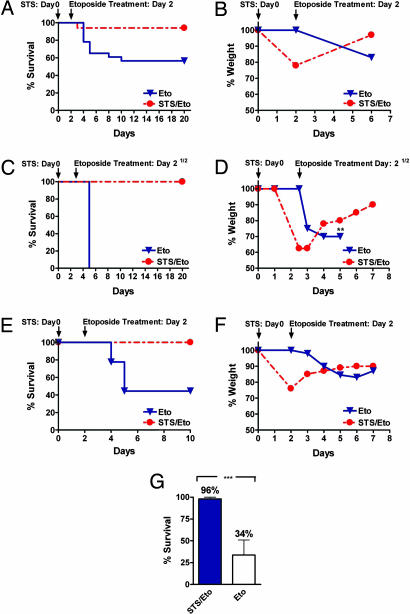
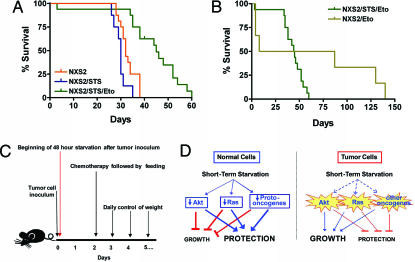
Strategies to treat cancer have focused primarily on the killing of tumor cells. Here, we describe a differential stress resistance (DSR) method that focuses instead on protecting the organism but not cancer cells against chemotherapy. Short-term starved S. cerevisiae or cells lacking proto-oncogene homologs were up to 1,000 times better protected against oxidative stress or chemotherapy drugs than cells expressing the oncogene homolog Ras2(val19). Low-glucose or low-serum media also protected primary glial cells but not six different rat and human glioma and neuroblastoma cancer cell lines against hydrogen peroxide or the chemotherapy drug/pro-oxidant cyclophosphamide. Finally, short-term starvation provided complete protection to mice but not to injected neuroblastoma cells against a high dose of the chemotherapy drug/pro-oxidant etoposide. These studies describe a starvation-based DSR strategy to enhance the efficacy of chemotherapy and suggest that specific agents among those that promote oxidative stress and DNA damage have the potential to maximize the differential toxicity to normal and cancer cells.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Could starvation minimize chemotherapy-induced toxicities?Expert Opin Ther Targets. 2008 Sep;12(9):1205-7. doi: 10.1517/14728222.12.9.1205. Expert Opin Ther Targets. 2008. PMID: 18694384
References
-
- Longo VD, Gralla EB, Valentine JS. J Biol Chem. 1996;271:12275–12280. - PubMed
-
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Science. 2001;292:288–290. - PubMed
-
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC, Cervera P, Le Bouc Y. Nature. 2003;421:182–187. - PubMed
-
- Lithgow GJ, White TM, Hinerfeld DA, Johnson TE. J Gerontol. 1994;49:B270–B276. - PubMed
-
- Longo VD, Finch CE. Science. 2003;299:1342–1346. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
