

The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits
- PMID: 18385101
- PMCID: PMC2700628
- DOI: 10.1093/hmg/ddn102
The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits
Abstract
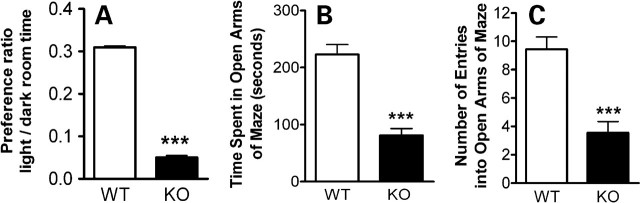
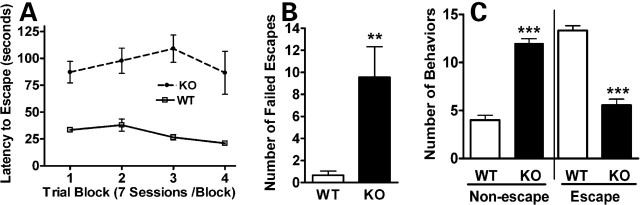
Methyl-CpG binding proteins (MBDs) are central components of DNA methylation-mediated epigenetic gene regulation. Alterations of epigenetic pathways are known to be associated with several neurodevelopmental disorders, particularly autism. Our previous studies showed that the loss of Mbd1 led to reduced hippocampal neurogenesis and impaired learning in mice. However, whether MBD1 regulates the autism-related cognitive functions remains unknown. Here we show that Mbd1 mutant (Mbd1(-/-)) mice exhibit several core deficits frequently associated with autism, including reduced social interaction, learning deficits, anxiety, defective sensory motor gating, depression and abnormal brain serotonin activity. Furthermore, we find that Mbd1 can directly regulate the expression of Htr2c, one of the serotonin receptors, by binding to its promoter, and the loss of Mbd1 led to elevated expression of Htr2c. Our results, therefore, demonstrate the importance of epigenetic regulation in mammalian brain development and cognitive functions. Understanding how the loss of Mbd1 could lead to autism-like behavioral phenotypes would reveal much-needed information about the molecular pathogenesis of autism.
Figures




References
-
- Muhle R., Trentacoste S.V., Rapin I. The genetics of autism. Pediatrics. 2004;113:e472–e486. - PubMed
-
- Holliday R., Pugh J.E. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. - PubMed
-
- Riggs A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell. Genet. 1975;14:9–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
