

Efficient control of population structure in model organism association mapping
- PMID: 18385116
- PMCID: PMC2278096
- DOI: 10.1534/genetics.107.080101
Efficient control of population structure in model organism association mapping
Abstract
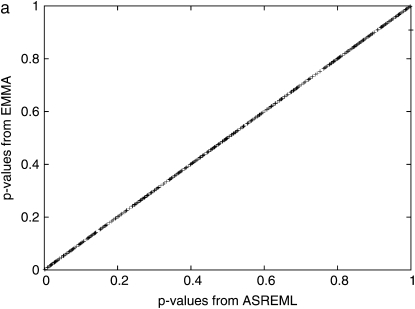
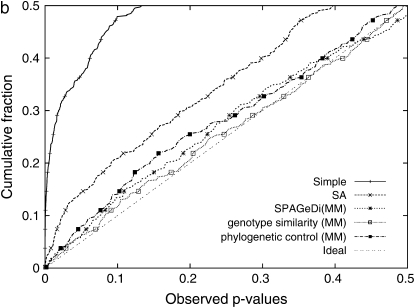
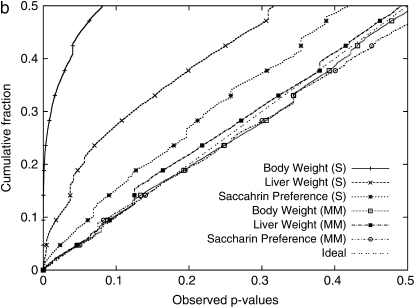
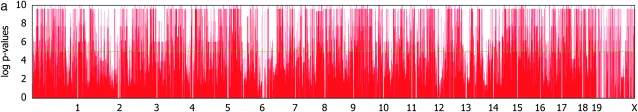
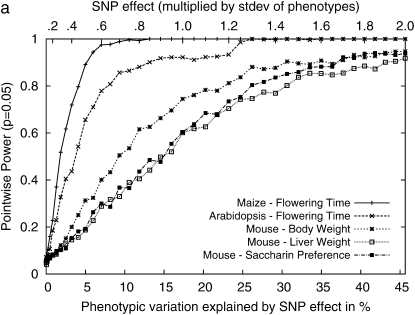
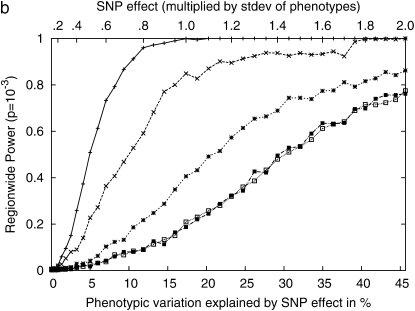
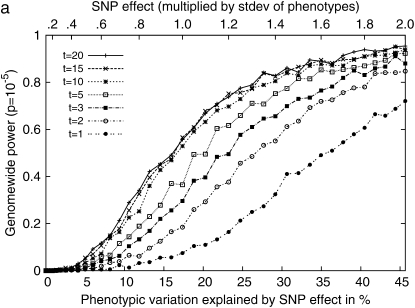
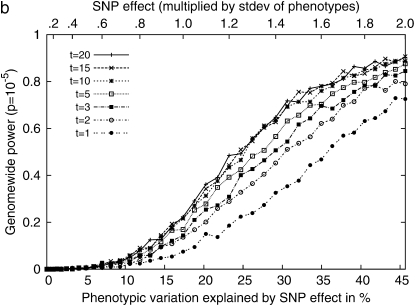
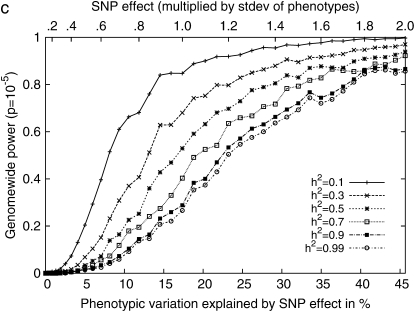
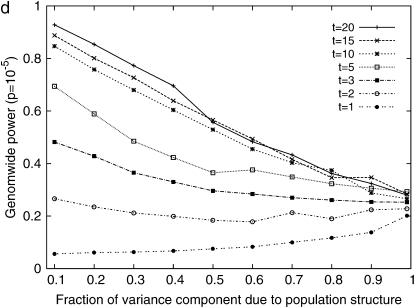
Genomewide association mapping in model organisms such as inbred mouse strains is a promising approach for the identification of risk factors related to human diseases. However, genetic association studies in inbred model organisms are confronted by the problem of complex population structure among strains. This induces inflated false positive rates, which cannot be corrected using standard approaches applied in human association studies such as genomic control or structured association. Recent studies demonstrated that mixed models successfully correct for the genetic relatedness in association mapping in maize and Arabidopsis panel data sets. However, the currently available mixed-model methods suffer from computational inefficiency. In this article, we propose a new method, efficient mixed-model association (EMMA), which corrects for population structure and genetic relatedness in model organism association mapping. Our method takes advantage of the specific nature of the optimization problem in applying mixed models for association mapping, which allows us to substantially increase the computational speed and reliability of the results. We applied EMMA to in silico whole-genome association mapping of inbred mouse strains involving hundreds of thousands of SNPs, in addition to Arabidopsis and maize data sets. We also performed extensive simulation studies to estimate the statistical power of EMMA under various SNP effects, varying degrees of population structure, and differing numbers of multiple measurements per strain. Despite the limited power of inbred mouse association mapping due to the limited number of available inbred strains, we are able to identify significantly associated SNPs, which fall into known QTL or genes identified through previous studies while avoiding an inflation of false positives. An R package implementation and webserver of our EMMA method are publicly available.
Figures

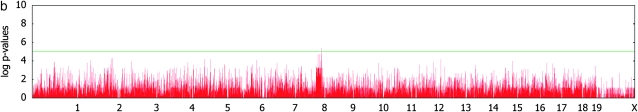
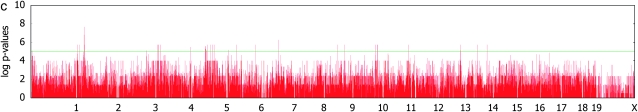
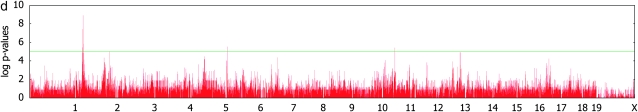
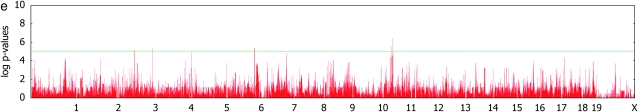

References
-
- Annuciado, R. V. P., M. Nishimura, M. Mori, A. Ishikawa, S. Tanaka et al., 2001. Quantitative trait loci for body weight in the intercross between SM/J and A/J mice. Exp. Anim. 50 319–324. - PubMed
-
- Arbelbide, M., J. Yu and R. Bernado, 2006. Power of mixed-model QTL mapping from phenotypic, pedigree and marker data in self-pollinated crops. Theor. Appl. Genet. 112 876–884. - PubMed
-
- Belknap, J. K., 1998. Effect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behav. Genet. 28 29–38. - PubMed
-
- Bhattacharya, T., M. Daniels, D. Heckerman, B. Foley, N. Frahm et al., 2007. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 315 1583–1586. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
