

IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release
- PMID: 18388325
- PMCID: PMC2430658
- DOI: 10.1161/CIRCRESAHA.108.173948
IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release
Erratum in
- Circ Res. 2009 Jul 2;105(1):e1
Abstract
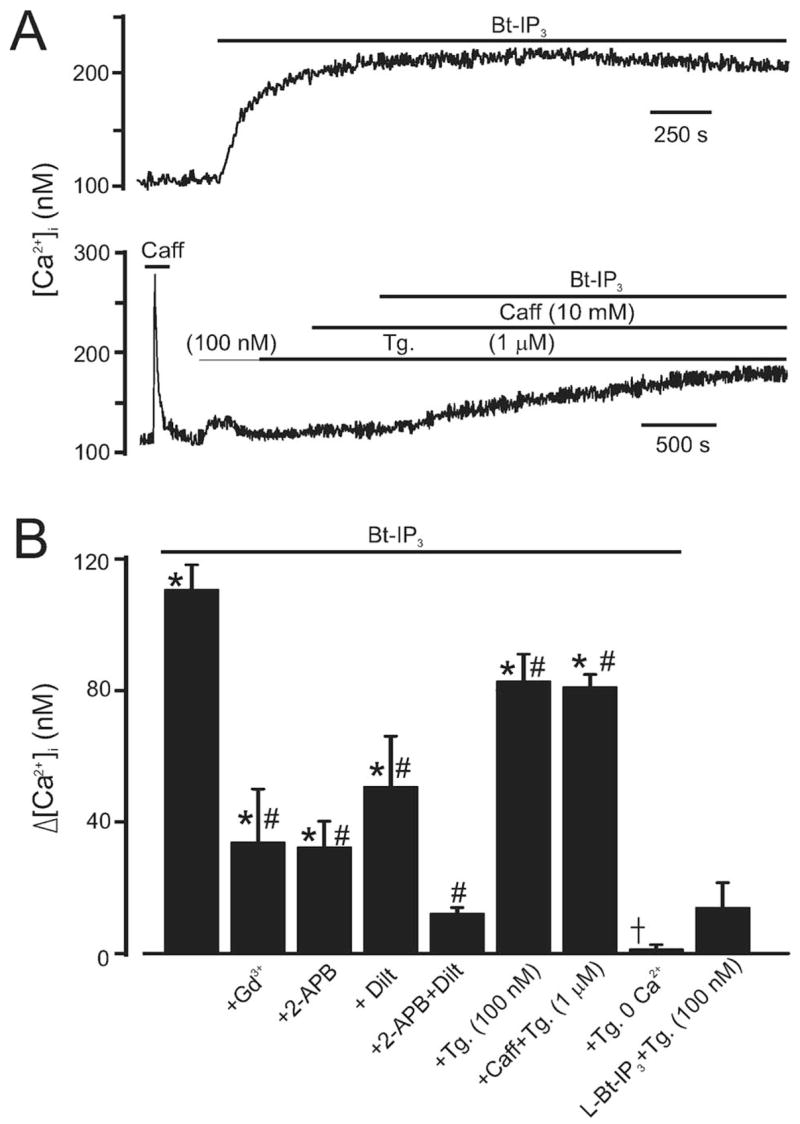
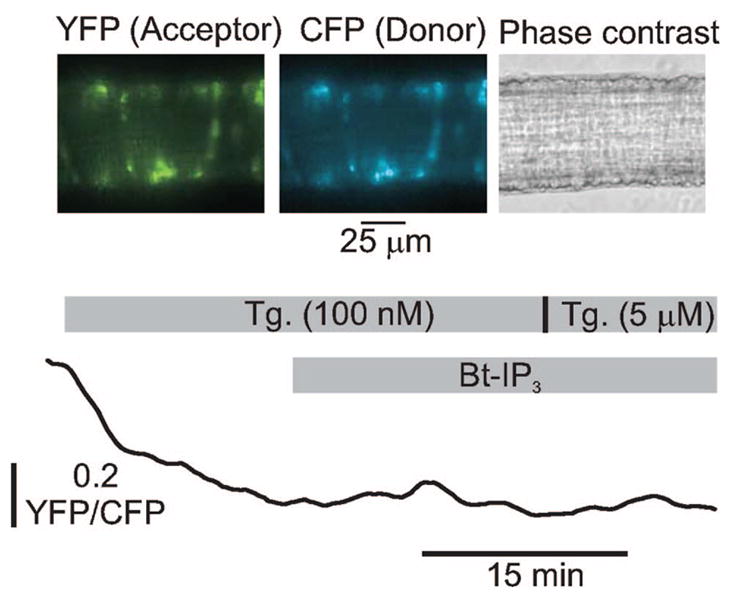
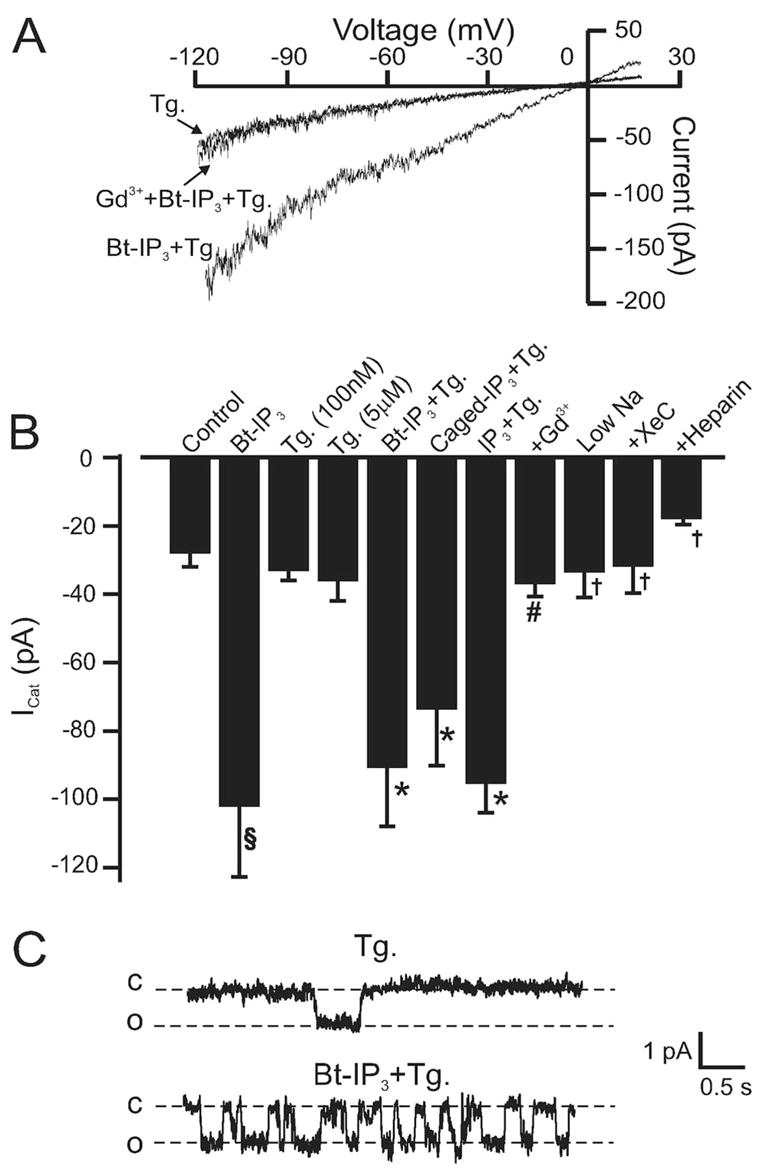
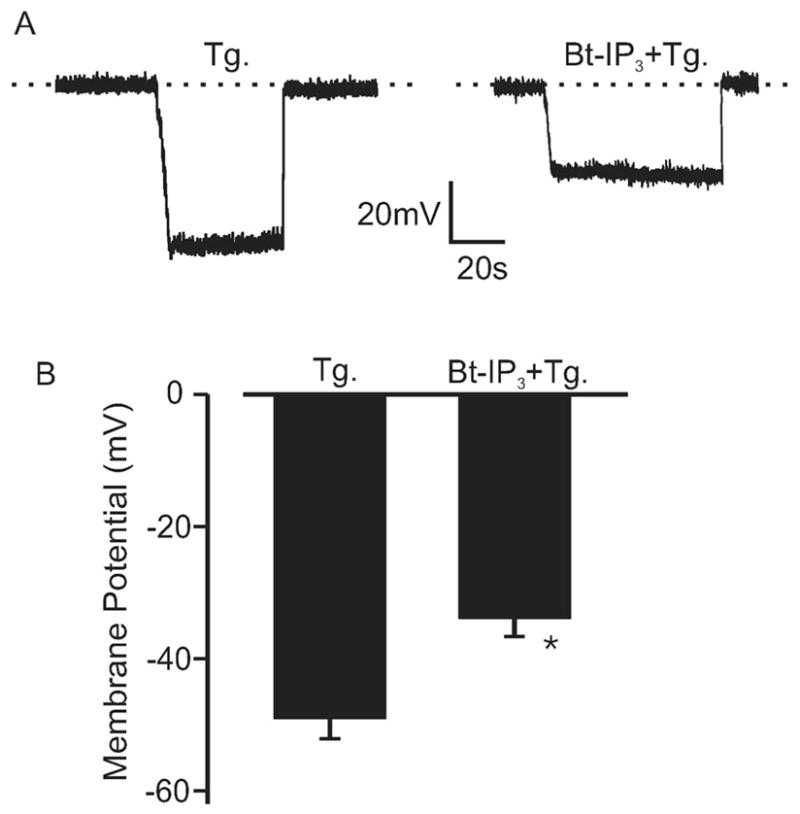
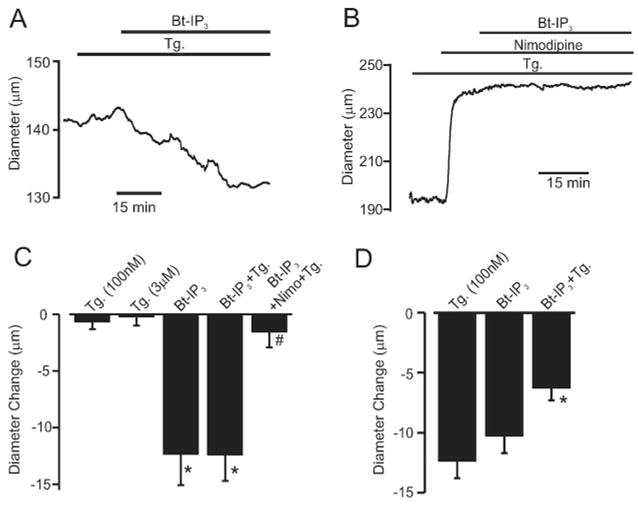
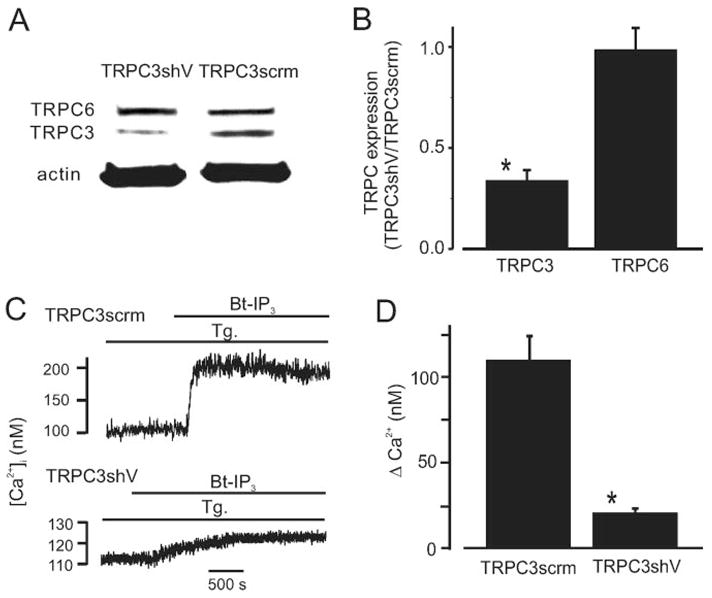
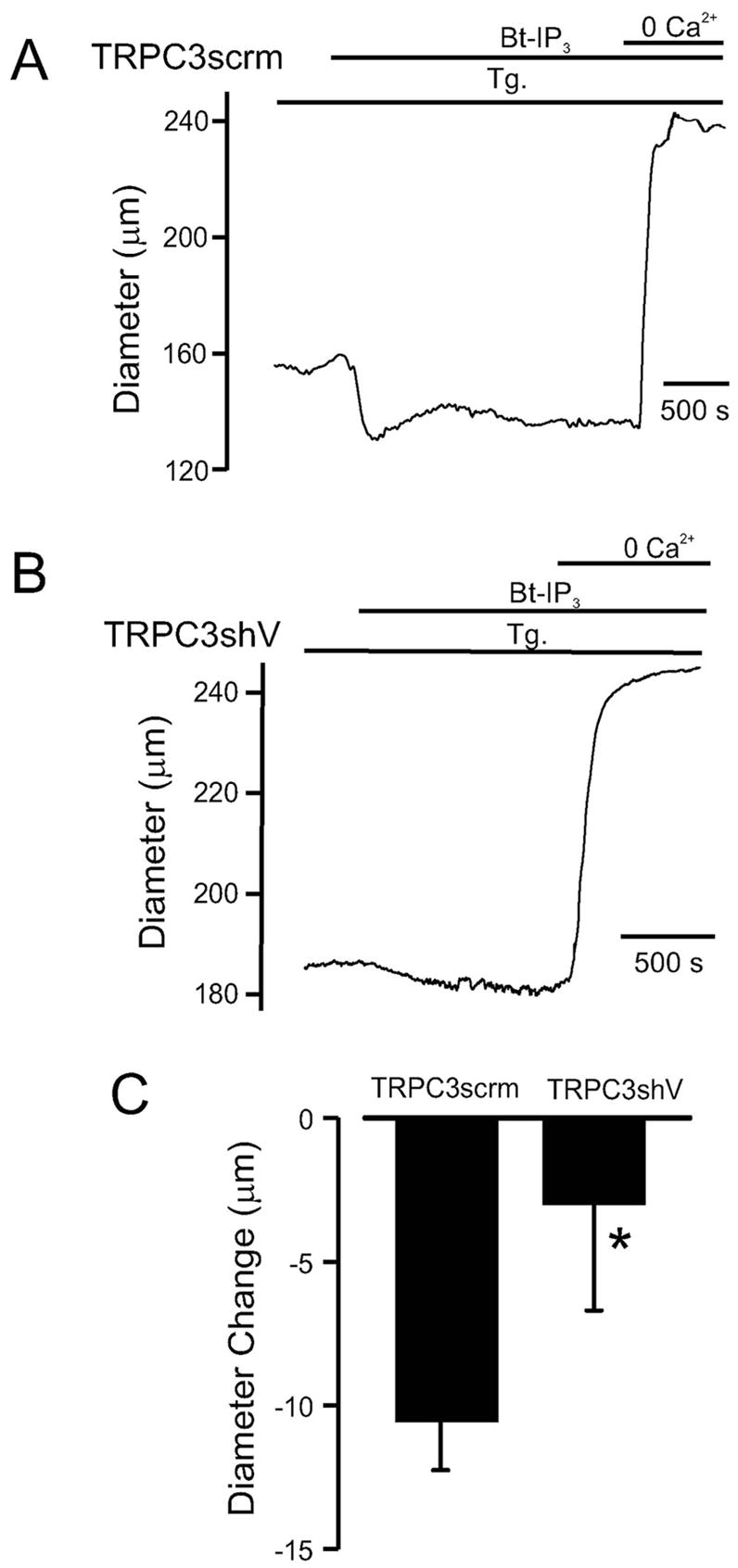
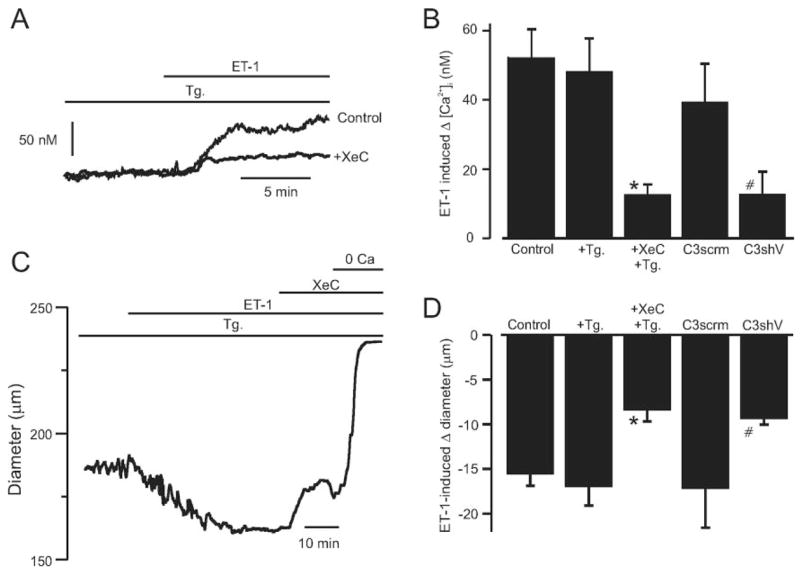
Vasoconstrictors that bind to phospholipase C-coupled receptors elevate inositol-1,4,5-trisphosphate (IP(3)). IP(3) is generally considered to elevate intracellular Ca(2+) concentration ([Ca(2+)](i)) in arterial myocytes and induce vasoconstriction via a single mechanism: by activating sarcoplasmic reticulum (SR)-localized IP(3) receptors, leading to intracellular Ca(2+) release. We show that IP(3) also stimulates vasoconstriction via a SR Ca(2+) release-independent mechanism. In isolated cerebral artery myocytes and arteries in which SR Ca(2+) was depleted to abolish Ca(2+) release (measured using D1ER, a fluorescence resonance energy transfer-based SR Ca(2+) indicator), IP(3) activated 15 pS sarcolemmal cation channels, generated a whole-cell cation current (I(Cat)) caused by Na(+) influx, induced membrane depolarization, elevated [Ca(2+)](i), and stimulated vasoconstriction. The IP(3)-induced I(Cat) and [Ca(2+)](i) elevation were attenuated by cation channel (Gd(3+), 2-APB) and IP(3) receptor (xestospongin C, heparin, 2-APB) blockers. TRPC3 (canonical transient receptor potential 3) channel knockdown with short hairpin RNA and diltiazem and nimodipine, voltage-dependent Ca(2+) channel blockers, reduced the SR Ca(2+) release-independent, IP(3)-induced [Ca(2+)](i) elevation and vasoconstriction. In pressurized arteries, SR Ca(2+) depletion did not alter IP(3)-induced constriction at 20 mm Hg but reduced IP(3)-induced constriction by approximately 39% at 60 mm Hg. [Ca(2+)](i) elevations and constrictions induced by endothelin-1, a phospholipase C-coupled receptor agonist, were both attenuated by TRPC3 knockdown and xestospongin C in SR Ca(2+)-depleted arteries. In summary, we describe a novel mechanism of IP(3)-induced vasoconstriction that does not occur as a result of SR Ca(2+) release but because of IP(3) receptor-dependent I(Cat) activation that requires TRPC3 channels. The resulting membrane depolarization activates voltage-dependent Ca(2+) channels, leading to a myocyte [Ca(2+)](i) elevation, and vasoconstriction.
Figures
References
-
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. - PubMed
-
- Clapham DE, Julius D, Montell C, Schultz G. International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev. 2005;57:427–450. - PubMed
-
- Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am J Physiol. 2001;281:C439–C448. - PubMed
-
- Dixit M, Zhuang D, Ceacareanu B, Hassid A. Treatment with insulin uncovers the motogenic capacity of nitric oxide in aortic smooth muscle cells: dependence on Gab1 and Gab1-SHP2 association. Circ Res. 2003;93:e113–e123. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
