

Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury
- PMID: 18391174
- PMCID: PMC3199571
- DOI: 10.1152/physrev.00024.2007
Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury
Abstract
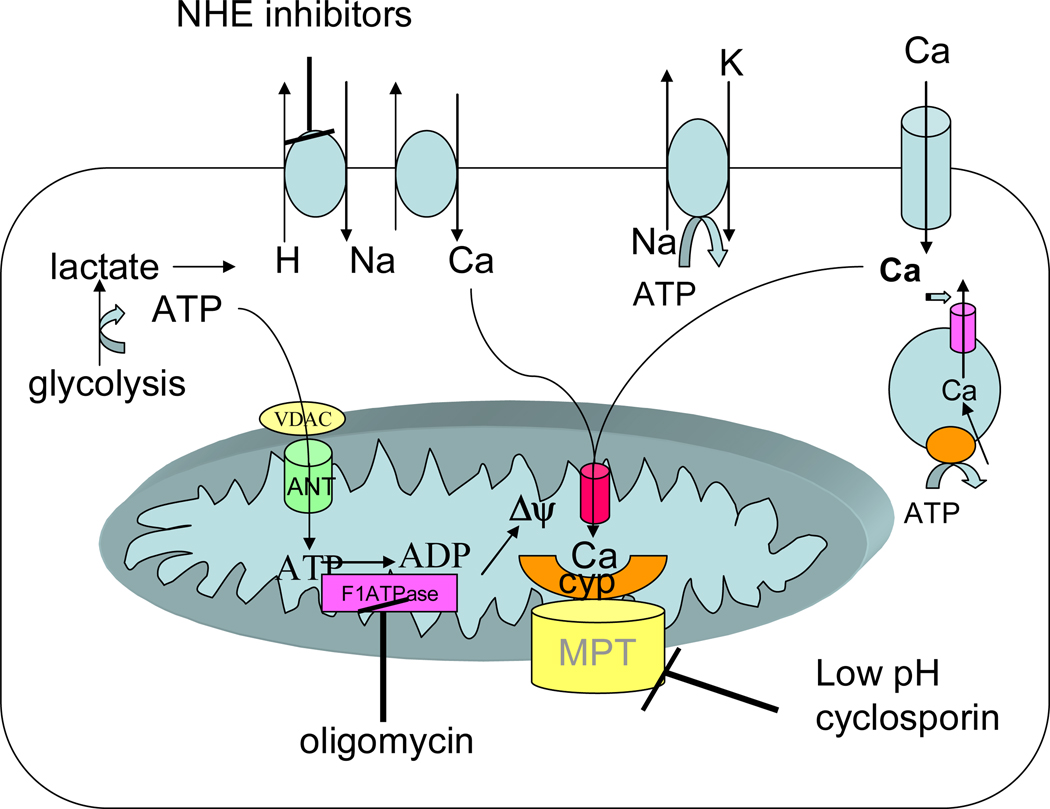
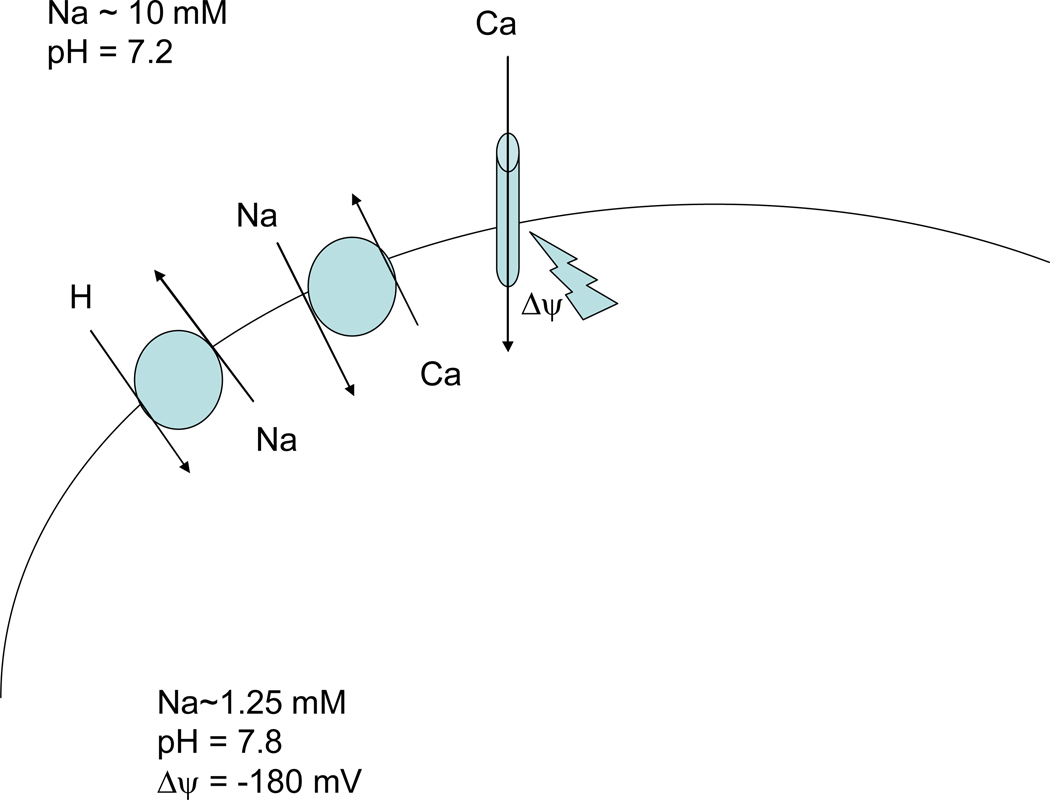
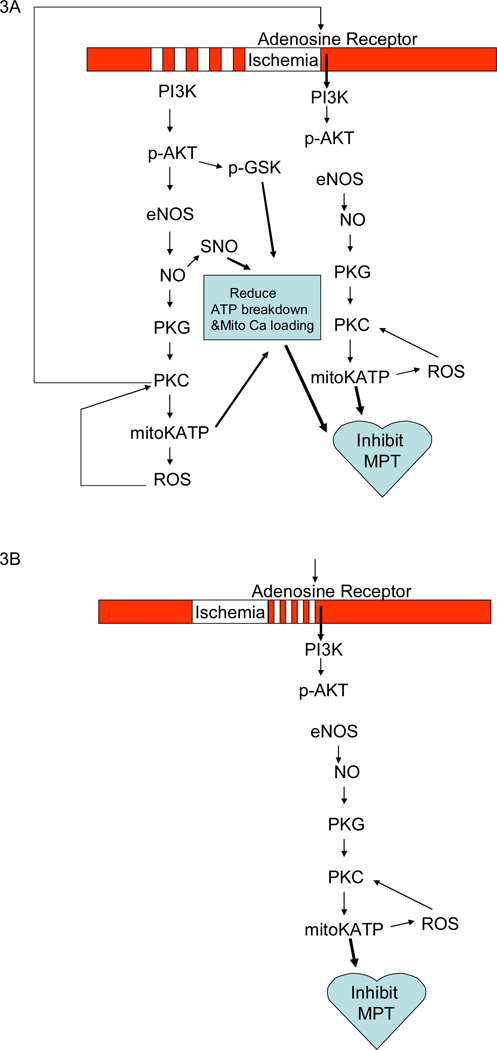
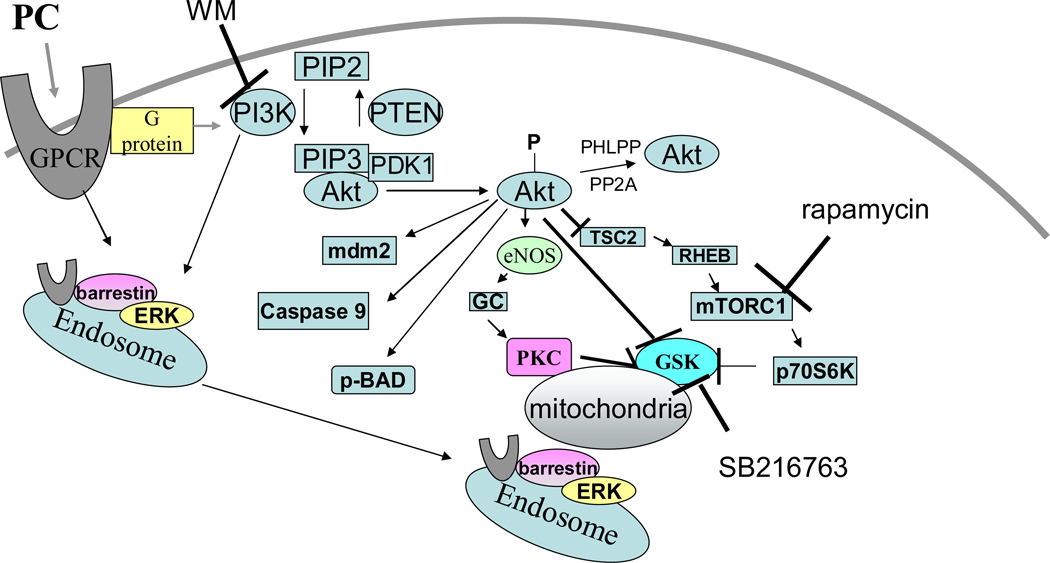
Mitochondria play an important role in cell death and cardioprotection. During ischemia, when ATP is progressively depleted, ion pumps cannot function resulting in a rise in calcium (Ca(2+)), which further accelerates ATP depletion. The rise in Ca(2+) during ischemia and reperfusion leads to mitochondrial Ca(2+) accumulation, particularly during reperfusion when oxygen is reintroduced. Reintroduction of oxygen allows generation of ATP; however, damage to the electron transport chain results in increased mitochondrial generation of reactive oxygen species (ROS). Mitochondrial Ca(2+) overload and increased ROS can result in opening of the mitochondrial permeability transition pore, which further compromises cellular energetics. The resultant low ATP and altered ion homeostasis result in rupture of the plasma membrane and cell death. Mitochondria have long been proposed as central players in cell death, since the mitochondria are central to synthesis of both ATP and ROS and since mitochondrial and cytosolic Ca(2+) overload are key components of cell death. Many cardioprotective mechanisms converge on the mitochondria to reduce cell death. Reducing Ca(2+) overload and reducing ROS have both been reported to reduce ischemic injury. Preconditioning activates a number of signaling pathways that reduce Ca(2+) overload and reduce activation of the mitochondrial permeability transition pore. The mitochondrial targets of cardioprotective signals are discussed in detail.
Figures
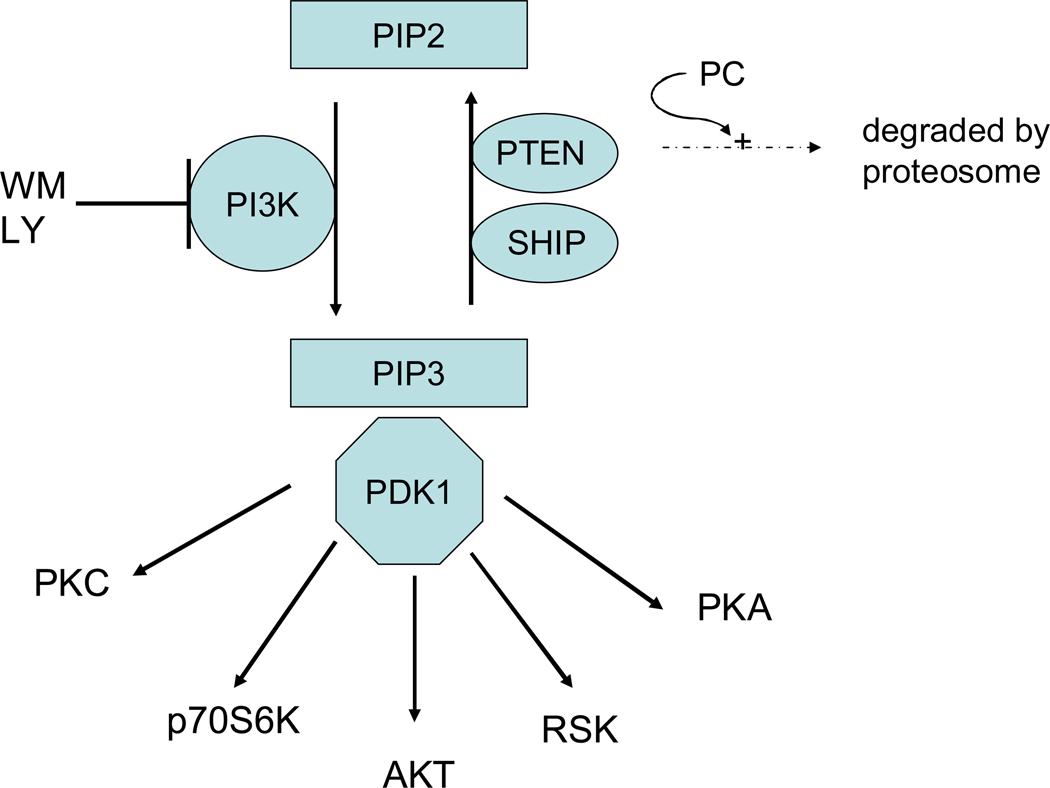
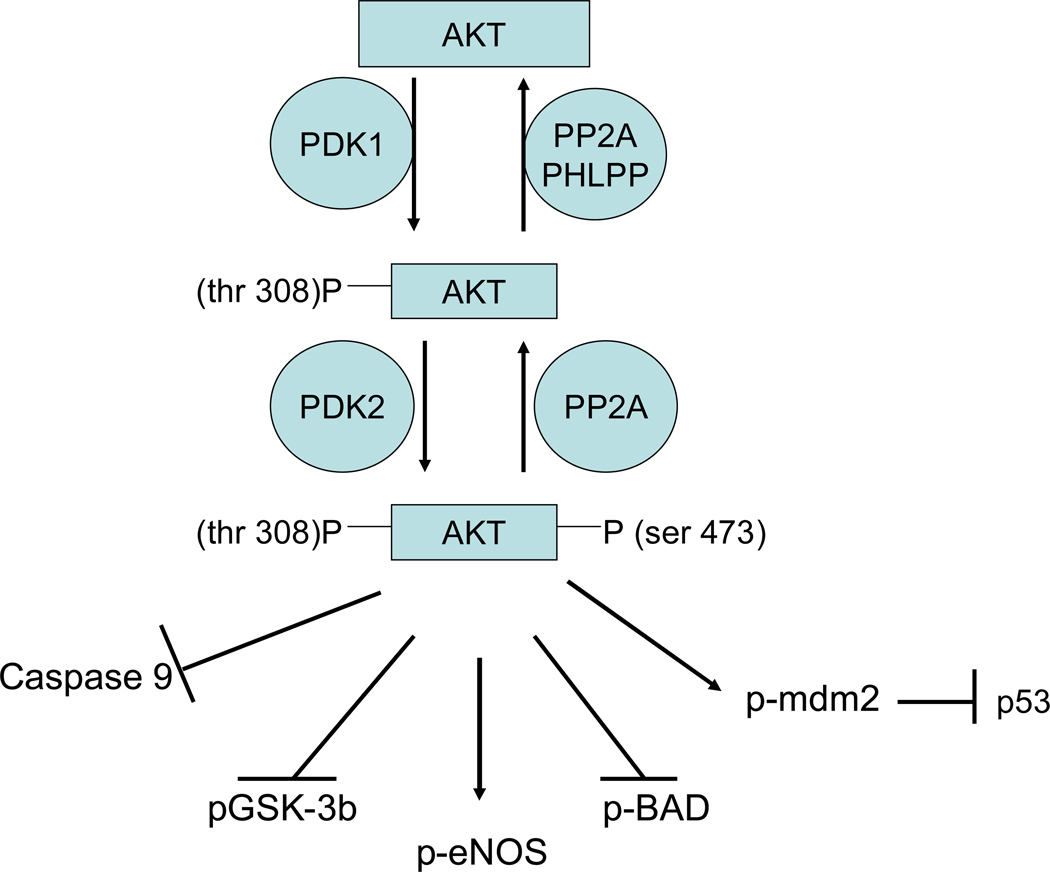
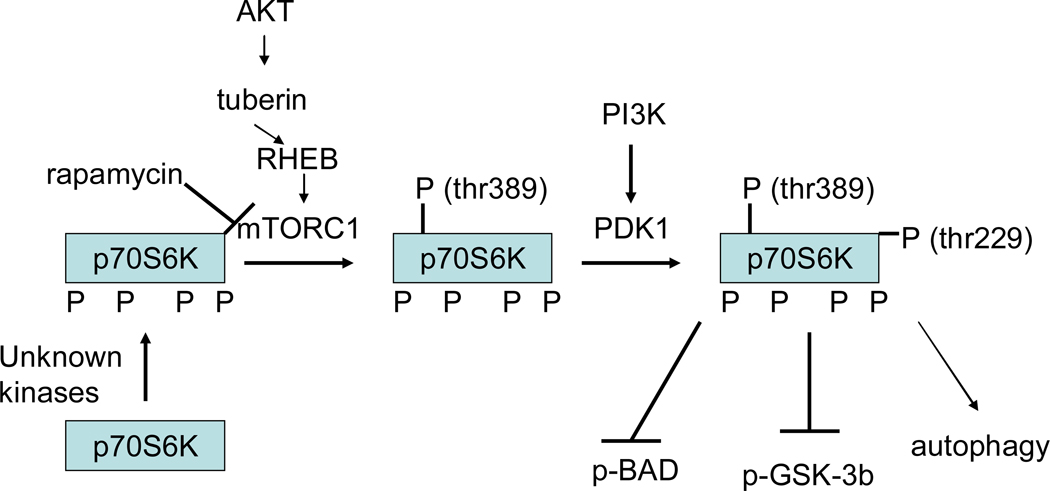
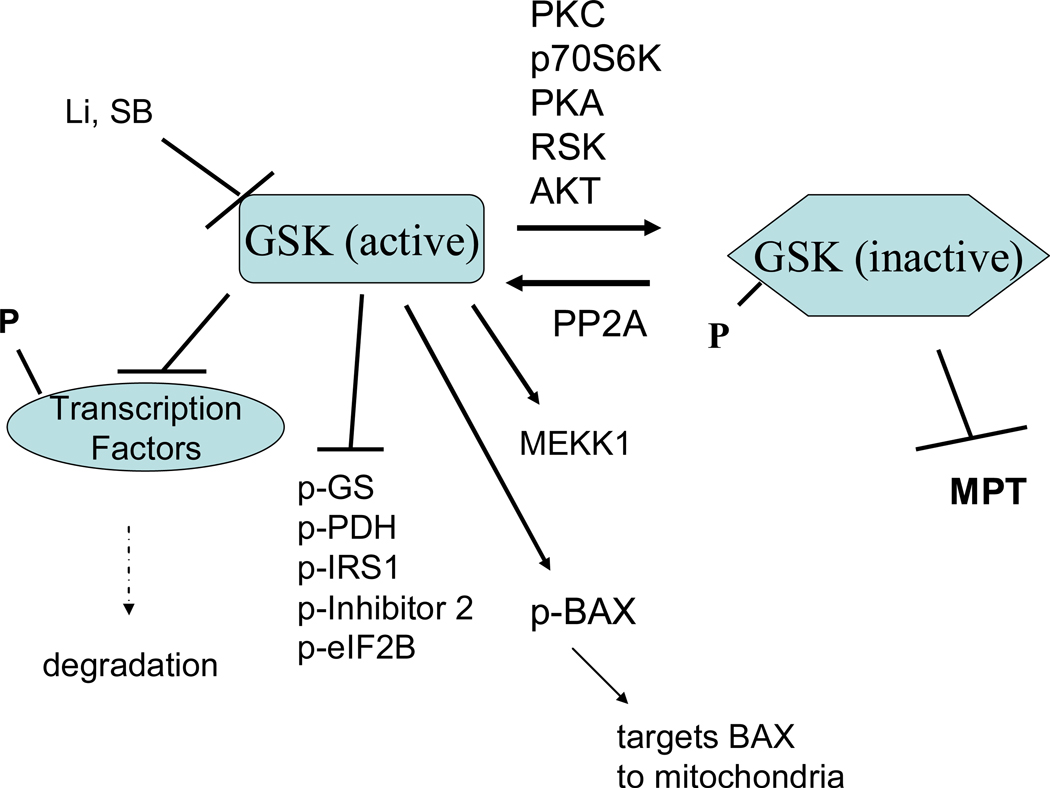
References
-
- MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:23–33. - PubMed
-
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. 2005;19:1088–1095. - PubMed
-
- Ahmad N, Wang Y, Haider KH, Wang B, Pasha Z, Uzun O, Ashraf M. Cardiac protection by mitoKATP channels is dependent on Akt translocation from cytosol to mitochondria during late preconditioning. Am J Physiol Heart Circ Physiol. 2006;290:H2402–H2408. - PubMed
-
- Ala-Rami A, Ylitalo KV, Hassinen IE. Ischaemic preconditioning and a mitochondrial KATP channel opener both produce cardioprotection accompanied by F1F0-ATPase inhibition in early ischaemia. Basic Res Cardiol. 2003;98:250–258. - PubMed
-
- Ali SM, Sabatini DM. Structure of S6 kinase 1 determines whether raptor-mTOR or rictor-mTOR phosphorylates its hydrophobic motif site. J Biol Chem. 2005;280:19445–19448. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous