

TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis
- PMID: 18396105
- PMCID: PMC3546119
- DOI: 10.1016/S1474-4422(08)70071-1
TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis
Abstract
Background: TDP-43 is a major component of the ubiquitinated inclusions that characterise amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) with ubiquitin inclusions (FTLD-U). TDP-43 is an RNA-binding and DNA-binding protein that has many functions and is encoded by the TAR DNA-binding protein gene (TARDBP) on chromosome 1. Our aim was to investigate whether TARDBP is a candidate disease gene for familial ALS that is not associated with mutations in superoxide dismutase 1 (SOD1).
Methods: TARDBP was sequenced in 259 patients with ALS, FTLD, or both. We used TaqMan-based SNP genotyping to screen for the identified variants in control groups matched to two kindreds of patients for age and ethnic origin. Additional clinical, genetic, and pathological assessments were made in these two families.
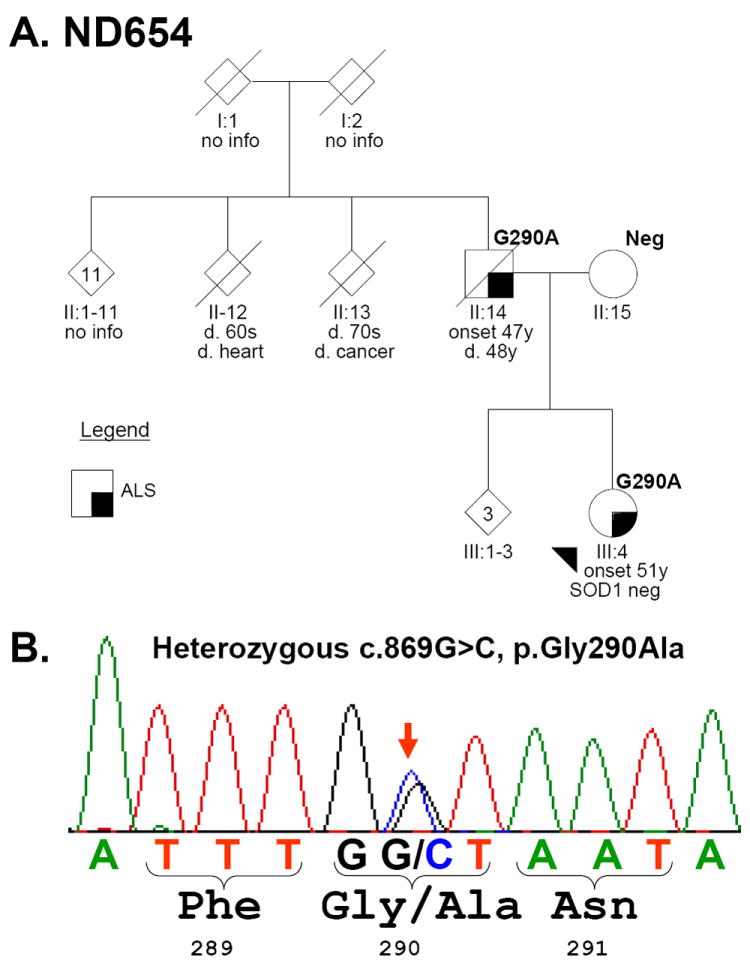
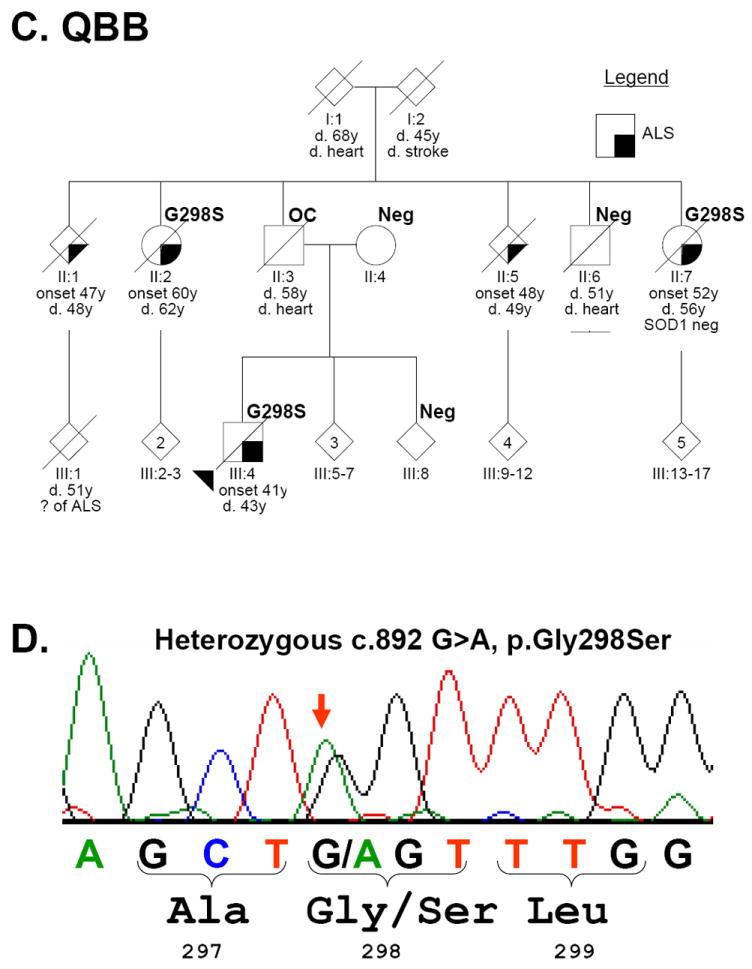
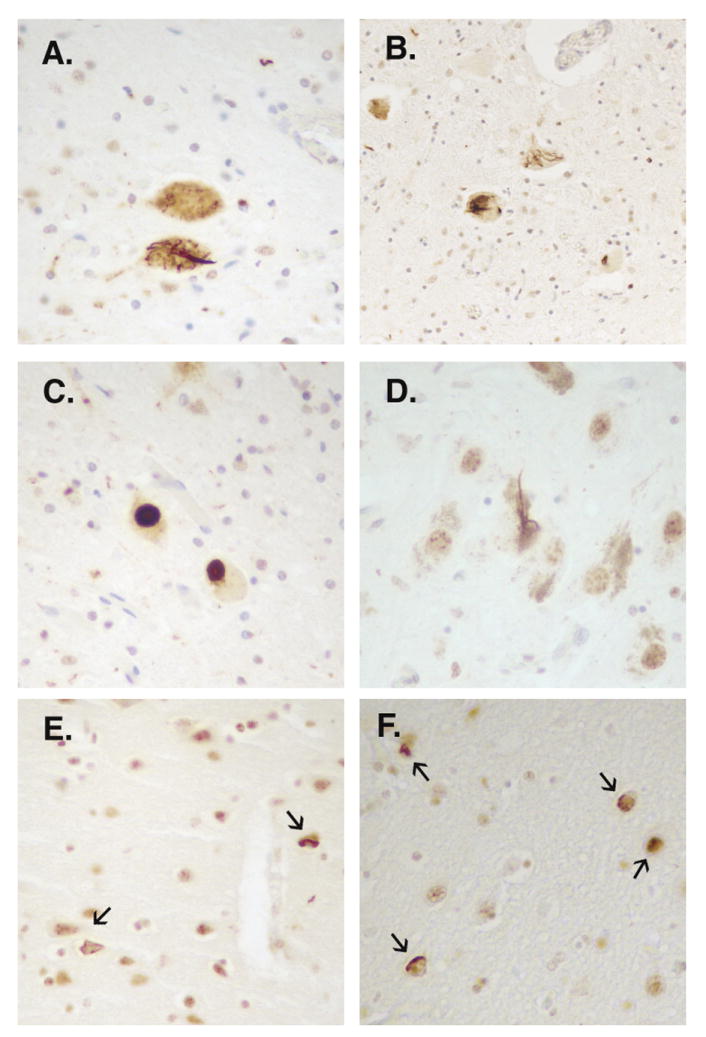
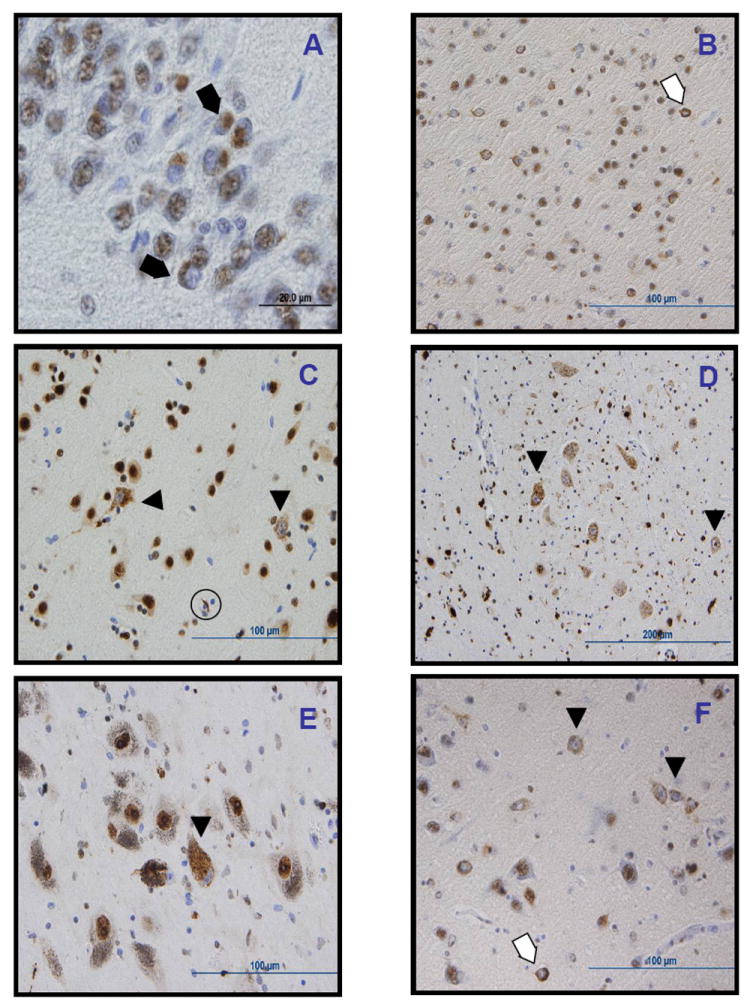
Findings: We identified two variants in TARDBP, which would encode Gly290Ala and Gly298Ser forms of TDP-43, in two kindreds with familial ALS. The variants seem to be pathogenic because they co-segregated with disease in both families, were absent in controls, and were associated with TDP-43 neuropathology in both members of one of these families for whom CNS tissue was available.
Interpretation: The Gly290Ala and Gly298Ser mutations are located in the glycine-rich domain of TDP-43, which regulates gene expression and mediates protein-protein interactions such as those with heterogeneous ribonucleoproteins. Owing to the varied and important cellular functions of TDP-43, these mutations might cause neurodegeneration through both gains and losses of function. The finding of pathogenic mutations in TARDBP implicates TDP-43 as an active mediator of neurodegeneration in TDP-43 proteinopathies, a class of disorder that includes ALS and FTLD-U.
Funding: National Institutes of Health (AG10124, AG17586, AG005136-22, PO1 AG14382), Department of Veterans Affairs, Friedrich-Baur Stiftung (0017/2007), US Public Health Service, ALS Association, and Fundació 'la Caixa'.
Figures
References
-
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. - PubMed
-
- Gros-Louis F, Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762(11-12):956–72. - PubMed
-
- Hadano S, Hand CK, Osuga H, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29(2):166–73. - PubMed
-
- Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29(2):160–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
