

A sensitized mutagenesis screen identifies Gli3 as a modifier of Sox10 neurocristopathy
- PMID: 18397875
- PMCID: PMC2902284
- DOI: 10.1093/hmg/ddn110
A sensitized mutagenesis screen identifies Gli3 as a modifier of Sox10 neurocristopathy
Abstract
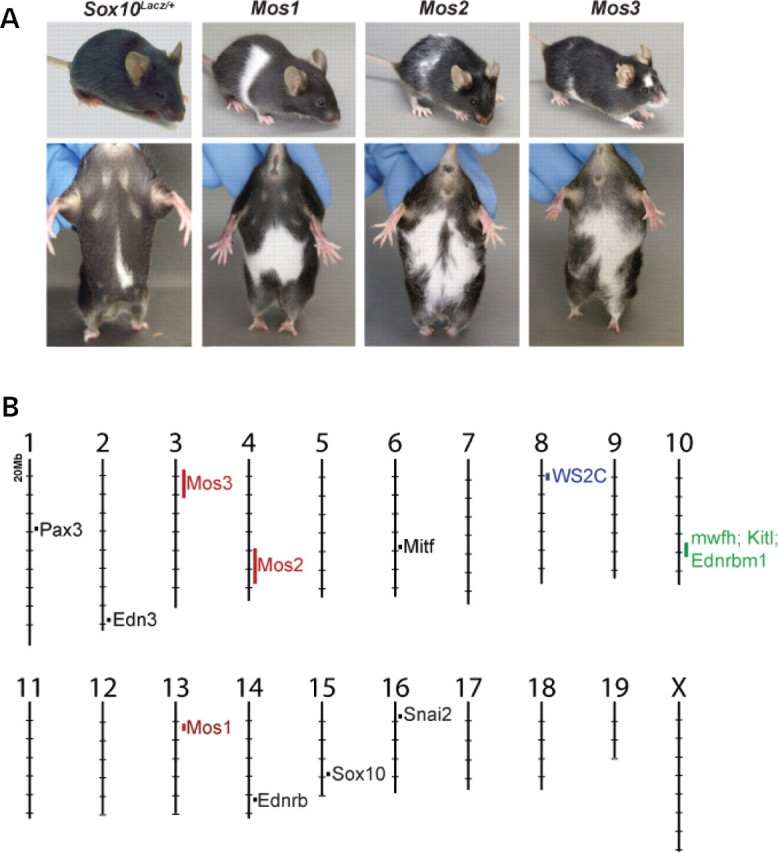
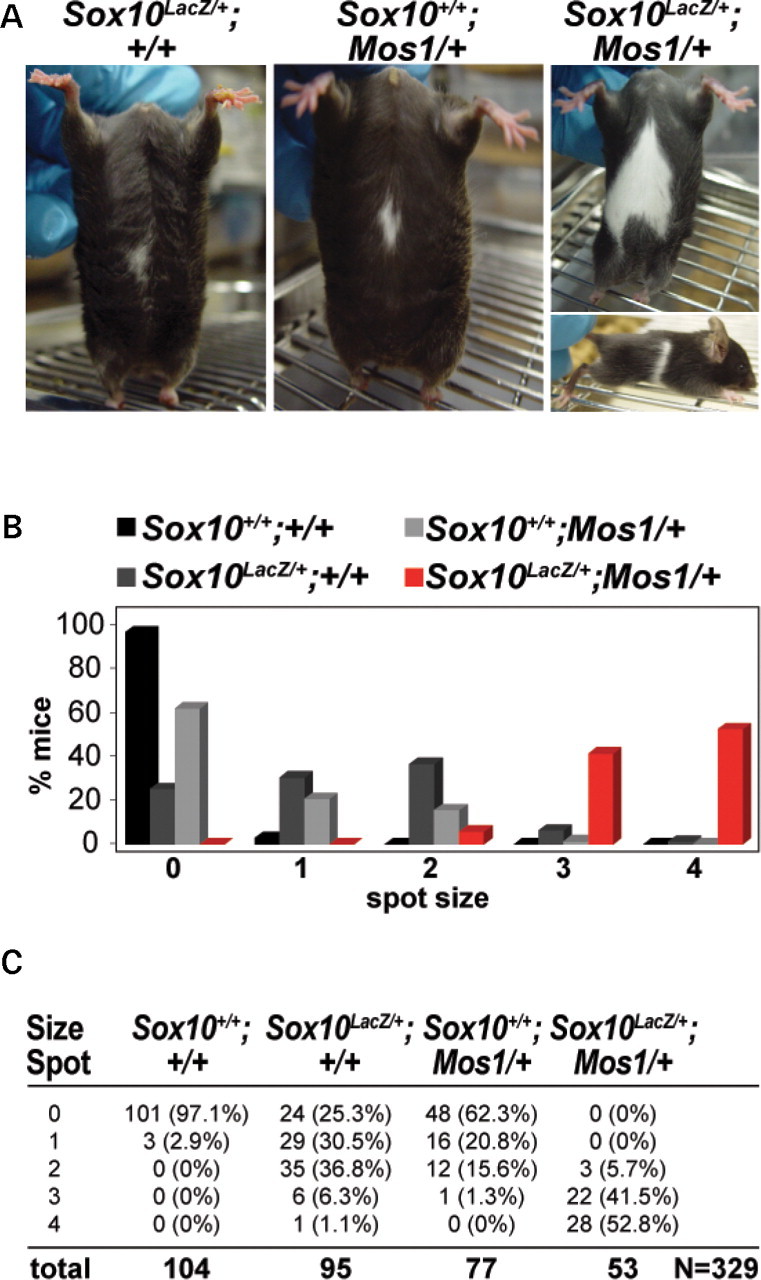
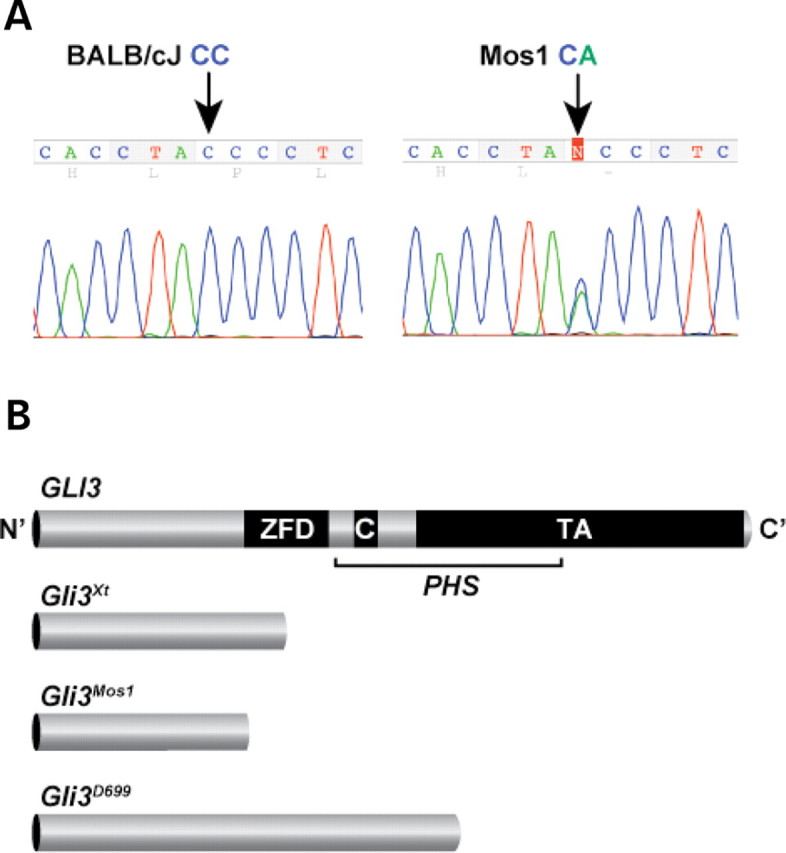
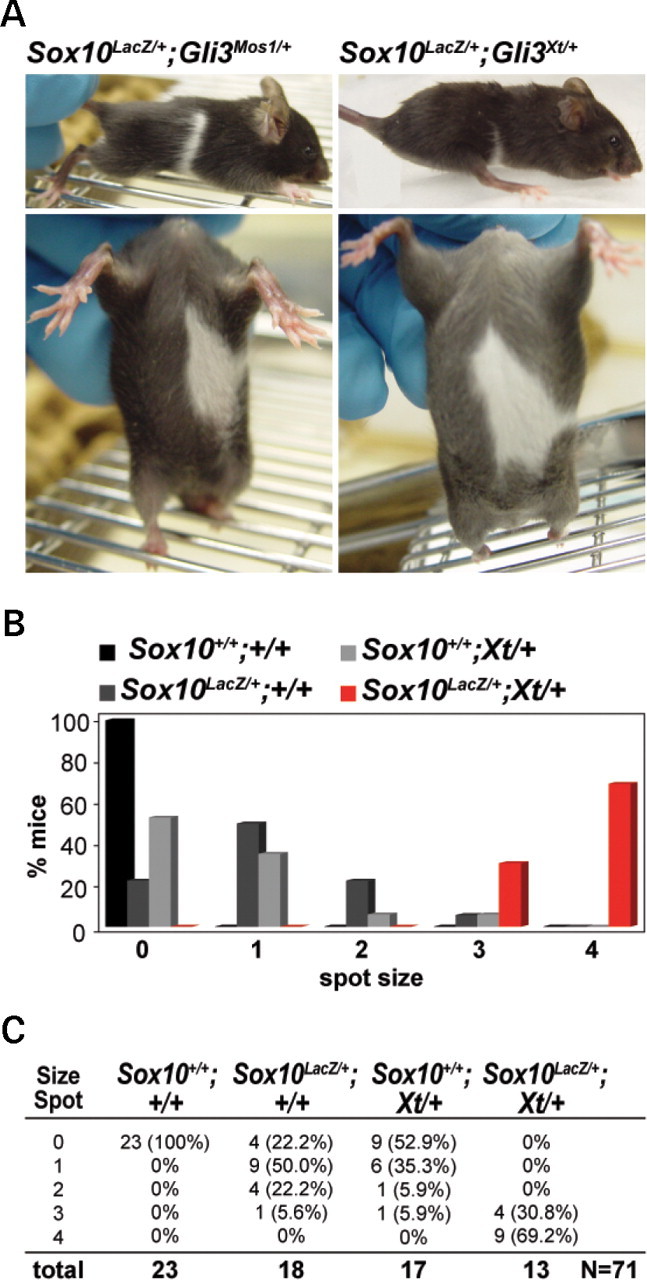
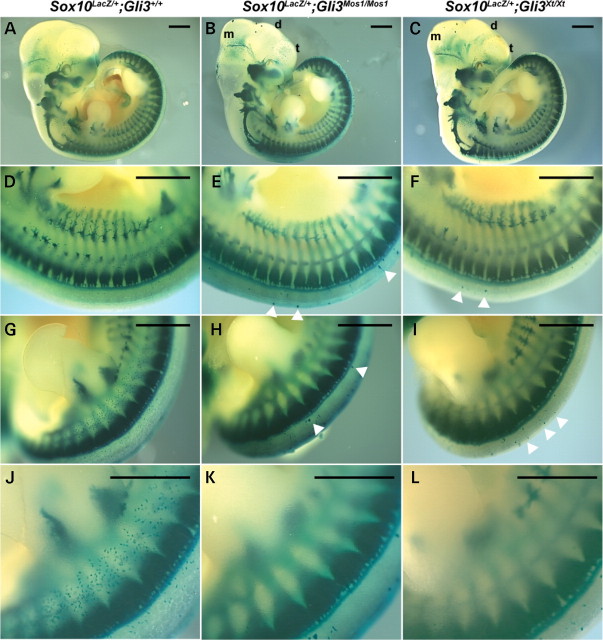
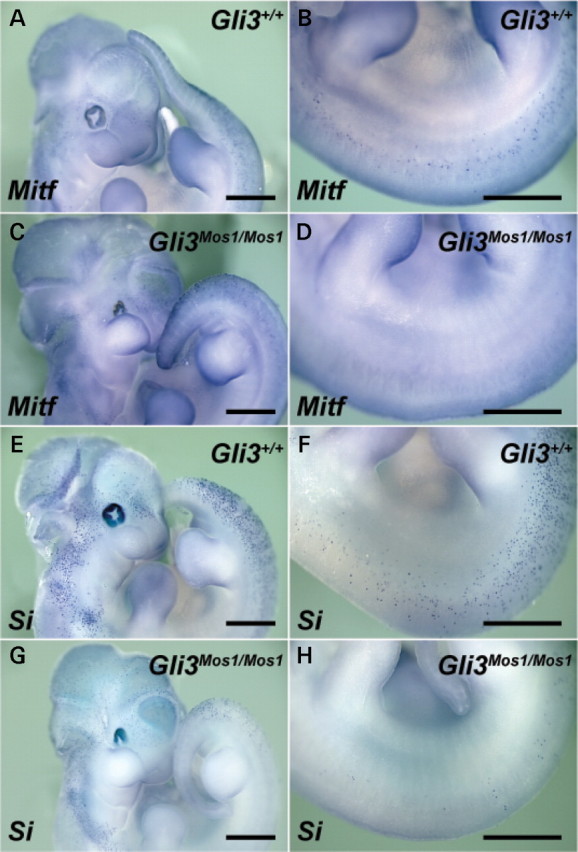
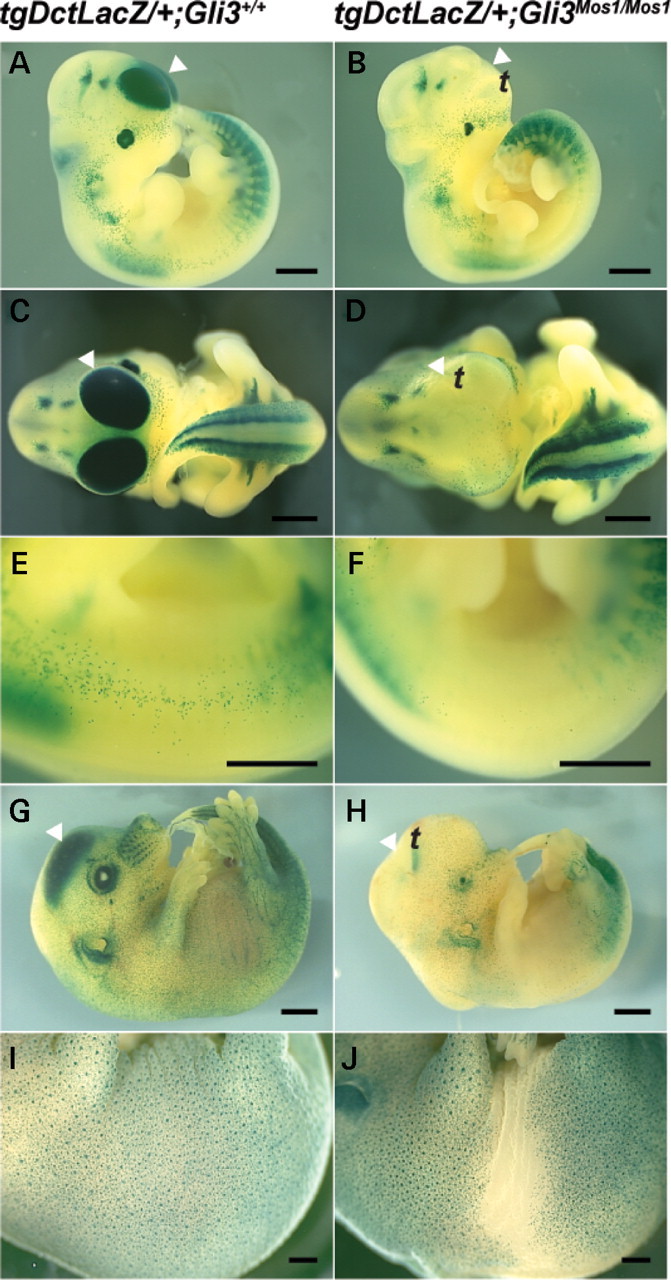
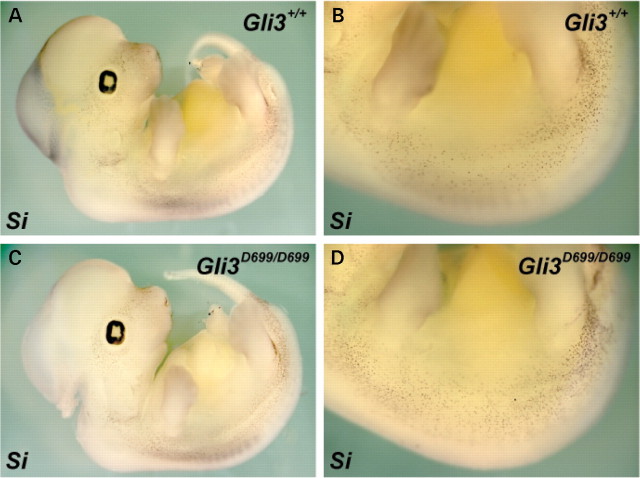
Haploinsufficiency for the transcription factor SOX10 is associated with the pigmentary deficiencies of Waardenburg syndrome (WS) and is modeled in Sox10 haploinsufficient mice (Sox10(LacZ/+)). As genetic background affects WS severity in both humans and mice, we established an N-ethyl-N-nitrosourea (ENU) mutagenesis screen to identify modifiers that increase the phenotypic severity of Sox10(LacZ/+) mice. Analysis of 230 pedigrees identified three modifiers, named modifier of Sox10 neurocristopathies (Mos1, Mos2 and Mos3). Linkage analysis confirmed their locations on mouse chromosomes 13, 4 and 3, respectively, within regions distinct from previously identified WS loci. Positional candidate analysis of Mos1 identified a truncation mutation in a hedgehog(HH)-signaling mediator, GLI-Kruppel family member 3 (Gli3). Complementation tests using a second allele of Gli3 (Gli3(Xt-J)) confirmed that a null mutation of Gli3 causes the increased hypopigmentation in Sox10(LacZ/+);Gli3(Mos1/)(+) double heterozygotes. Early melanoblast markers (Mitf, Sox10, Dct, and Si) are reduced in Gli3(Mos1/)(Mos1) embryos, indicating that loss of GLI3 signaling disrupts melanoblast specification. In contrast, mice expressing only the GLI3 repressor have normal melanoblast specification, indicating that the full-length GLI3 activator is not required for specification of neural crest to the melanocyte lineage. This study demonstrates the feasibility of sensitized screens to identify disease modifier loci and implicates GLI3 and other HH signaling components as modifiers of human neurocristopathies.
Figures
Similar articles
-
A Sox10 expression screen identifies an amino acid essential for Erbb3 function.PLoS Genet. 2008 Sep 5;4(9):e1000177. doi: 10.1371/journal.pgen.1000177. PLoS Genet. 2008. PMID: 18773073 Free PMC article.
-
Genetic evidence does not support direct regulation of EDNRB by SOX10 in migratory neural crest and the melanocyte lineage.Mech Dev. 2006 Feb;123(2):124-34. doi: 10.1016/j.mod.2005.11.004. Epub 2006 Jan 18. Mech Dev. 2006. PMID: 16412618 Free PMC article.
-
Identification of GLI Mutations in Patients With Hirschsprung Disease That Disrupt Enteric Nervous System Development in Mice.Gastroenterology. 2015 Dec;149(7):1837-1848.e5. doi: 10.1053/j.gastro.2015.07.060. Epub 2015 Aug 7. Gastroenterology. 2015. PMID: 26261006
-
The importance of having your SOX on: role of SOX10 in the development of neural crest-derived melanocytes and glia.Oncogene. 2003 May 19;22(20):3024-34. doi: 10.1038/sj.onc.1206442. Oncogene. 2003. PMID: 12789277 Review.
-
Sorting out Sox10 functions in neural crest development.Bioessays. 2006 Aug;28(8):788-98. doi: 10.1002/bies.20445. Bioessays. 2006. PMID: 16927299 Review.
Cited by
-
Disrupting Hedgehog signaling in melanocytes by SUFU knockout leads to ocular melanocytosis and anterior segment malformation.Dis Model Mech. 2023 Aug 1;16(8):dmm050210. doi: 10.1242/dmm.050210. Epub 2023 Aug 29. Dis Model Mech. 2023. PMID: 37577930 Free PMC article.
-
MMTV-Wnt1 and -DeltaN89beta-catenin induce canonical signaling in distinct progenitors and differentially activate Hedgehog signaling within mammary tumors.PLoS One. 2009;4(2):e4537. doi: 10.1371/journal.pone.0004537. Epub 2009 Feb 19. PLoS One. 2009. PMID: 19225568 Free PMC article.
-
Towards better mouse models: enhanced genotypes, systemic phenotyping and envirotype modelling.Nat Rev Genet. 2009 Jun;10(6):371-80. doi: 10.1038/nrg2578. Nat Rev Genet. 2009. PMID: 19434078 Review.
-
The Genome of C57BL/6J "Eve", the Mother of the Laboratory Mouse Genome Reference Strain.G3 (Bethesda). 2019 Jun 5;9(6):1795-1805. doi: 10.1534/g3.119.400071. G3 (Bethesda). 2019. PMID: 30996023 Free PMC article.
-
BRG1 interacts with SOX10 to establish the melanocyte lineage and to promote differentiation.Nucleic Acids Res. 2017 Jun 20;45(11):6442-6458. doi: 10.1093/nar/gkx259. Nucleic Acids Res. 2017. PMID: 28431046 Free PMC article.
References
-
- Baldwin C.T., Hoth C.F., Amos J.A., da-Silva E.O., Milunsky A. An exonic mutation in the HuP2 paired domain gene causes Waardenburg’s syndrome. Nature. 1992;355:637–638. - PubMed
-
- Tassabehji M., Read A.P., Newton V.E., Harris R., Balling R., Gruss P., Strachan T. Waardenburg’s syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature. 1992;355:635–636. - PubMed
-
- Tassabehji M., Newton V.E., Read A.P. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat. Genet. 1994;8:251–255. - PubMed
-
- Sanchez-Martin M., Rodriguez-Garcia A., Perez-Losada J., Sagrera A., Read A.P., Sanchez-Garcia I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum. Mol. Genet. 2002;11:3231–3236. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
