

Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration
- PMID: 18400166
- PMCID: PMC2394729
- DOI: 10.1016/j.neuron.2008.01.033
Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration
Abstract
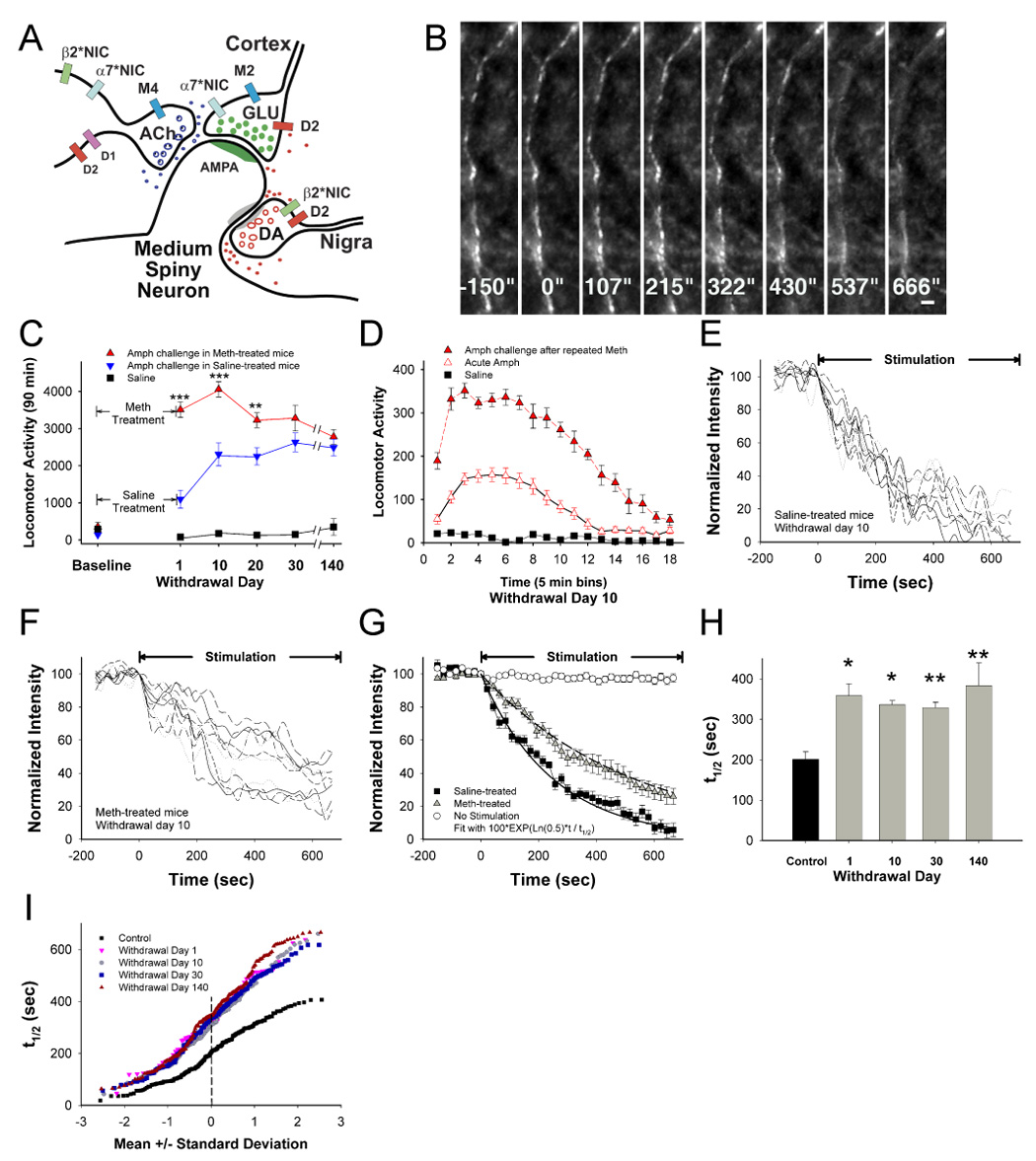
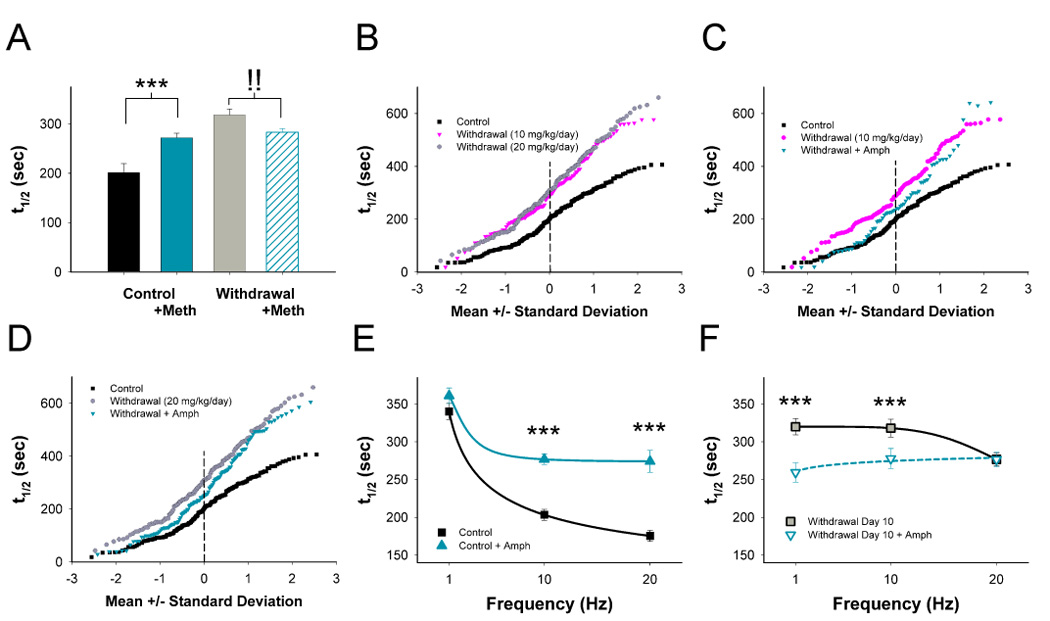
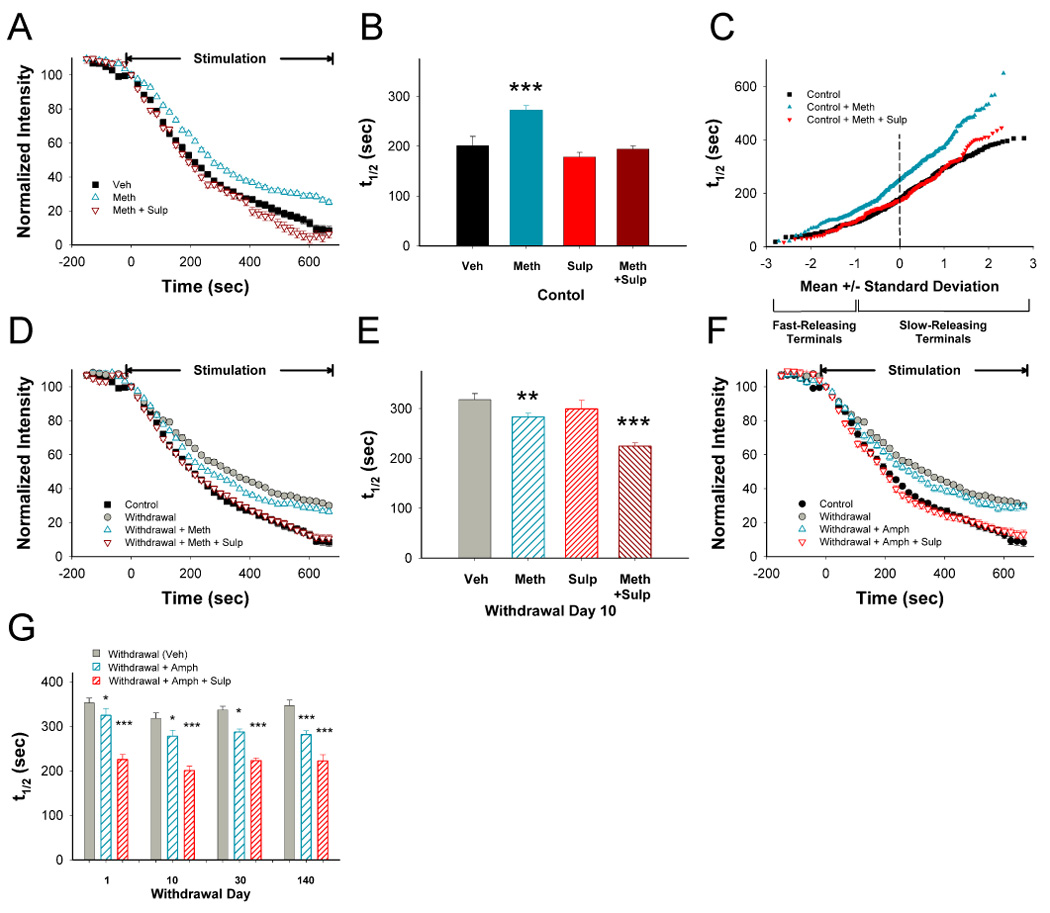
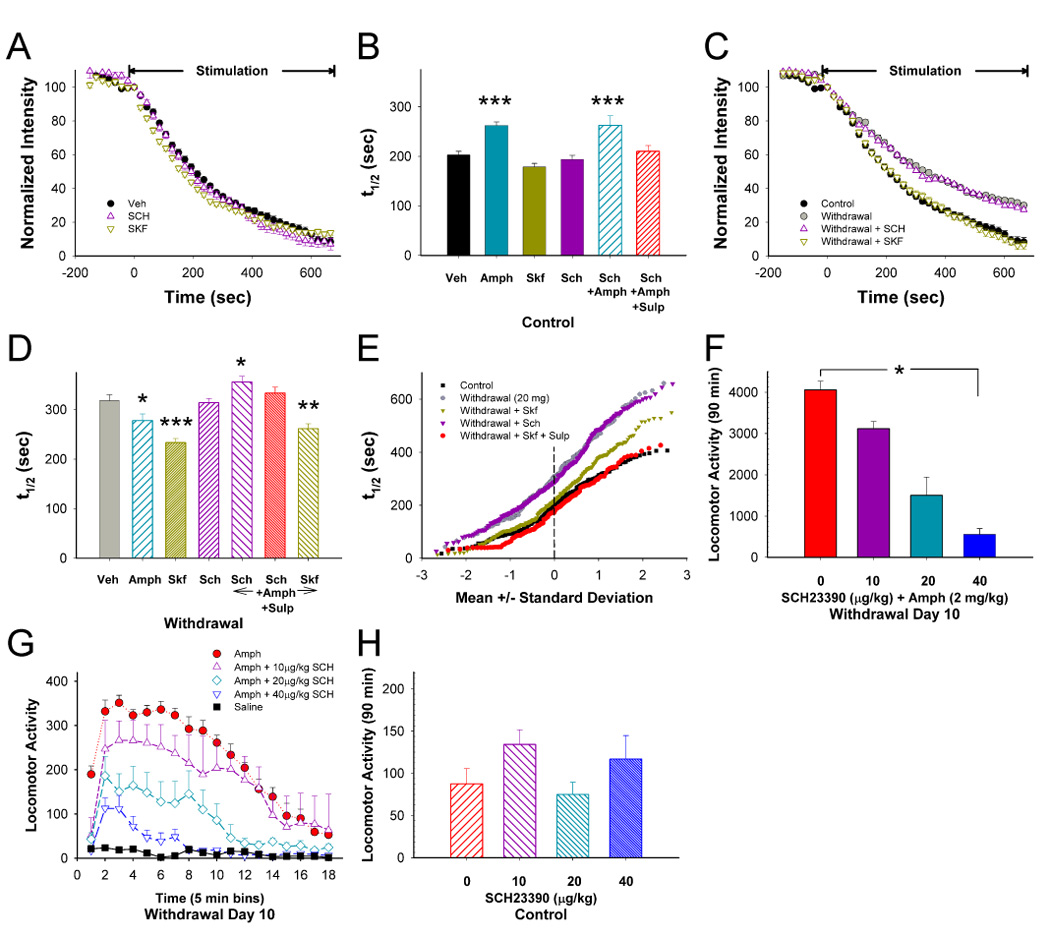
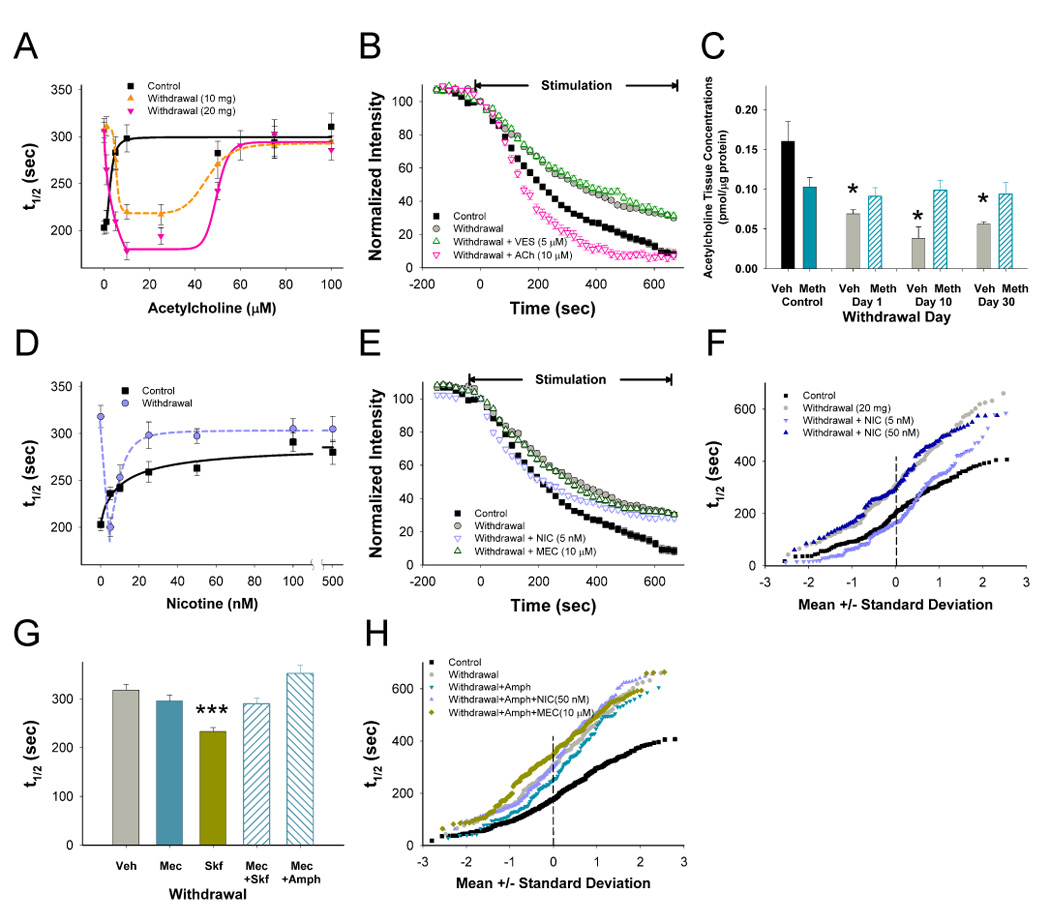
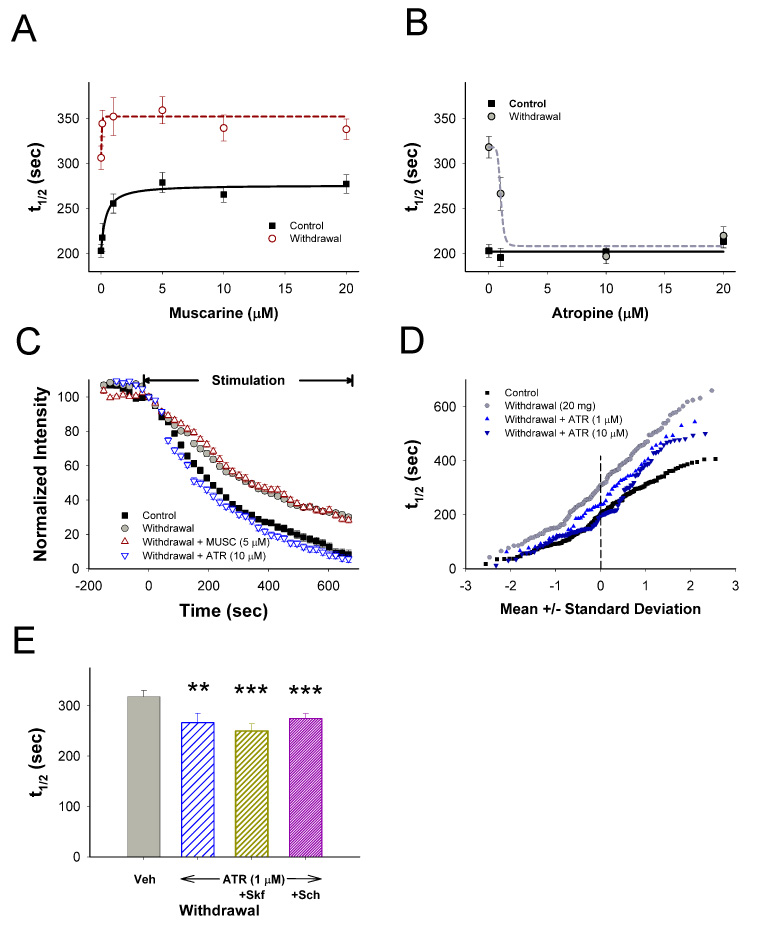
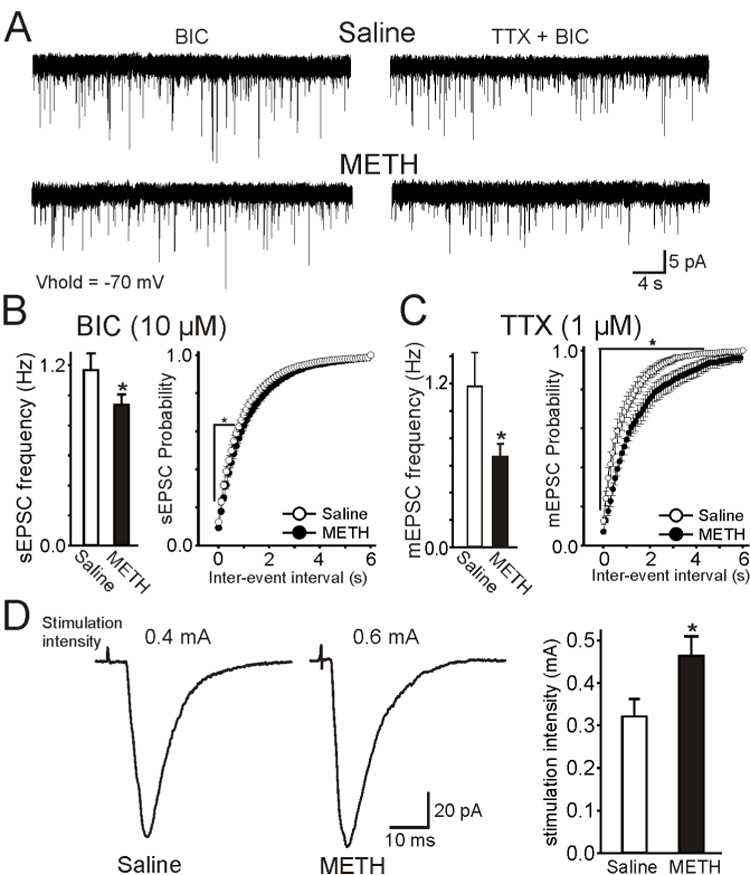
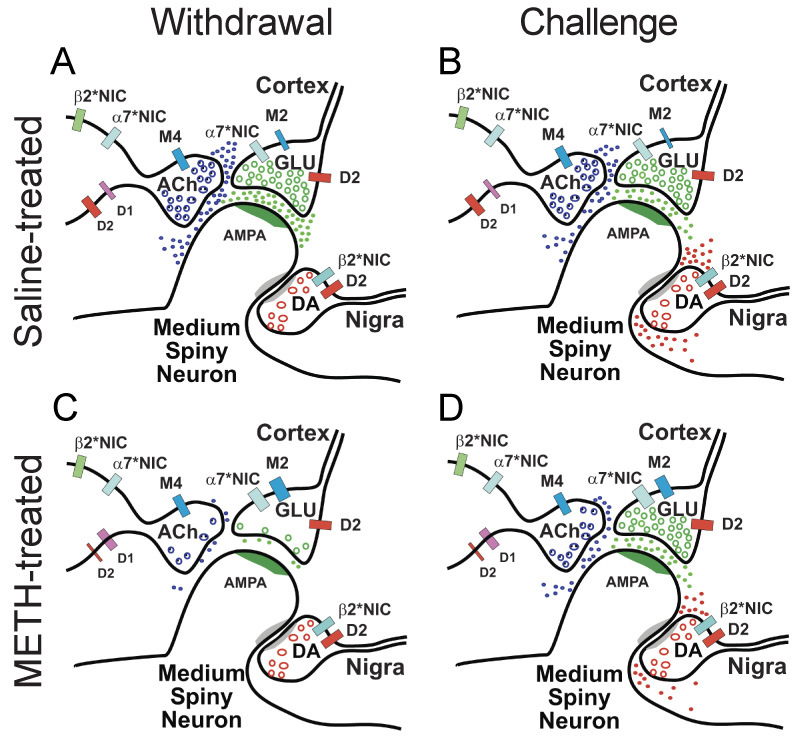
Addiction-associated behaviors such as drug craving and relapse are hypothesized to result from synaptic changes that persist long after withdrawal and are renormalized by drug reinstatement, although such chronic synaptic effects have not been identified. We report that exposure to the dopamine releaser methamphetamine for 10 days elicits a long-lasting (>4 month) depression at corticostriatal terminals that is reversed by methamphetamine readministration. Both methamphetamine-induced chronic presynaptic depression and the drug's selective renormalization in drug-experienced animals are independent of corresponding long-term changes in synaptic dopamine release but are due to alterations in D1 dopamine and cholinergic receptor systems. These mechanisms might provide a synaptic basis that underlies addiction and habit learning and their long-term maintenance.
Figures
Comment in
-
Methamphetamine induces chronic corticostriatal depression: too much of a bad thing.Neuron. 2008 Apr 10;58(1):6-7. doi: 10.1016/j.neuron.2008.03.014. Neuron. 2008. PMID: 18400156 Review.
References
-
- Ahmed SH, Koob GF. Transition to drug addiction: a negative reinforcement model based on an allostatic decrease in reward function. Psychopharmacology (Berl) 2005;180:473–490. - PubMed
-
- Azam L, Winzer-Serhan U, Leslie FM. Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience. 2003;119:965–977. - PubMed
-
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004b;42:653–663. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
