

GpnmbR150X allele must be present in bone marrow derived cells to mediate DBA/2J glaucoma
- PMID: 18402690
- PMCID: PMC2373794
- DOI: 10.1186/1471-2156-9-30
GpnmbR150X allele must be present in bone marrow derived cells to mediate DBA/2J glaucoma
Abstract
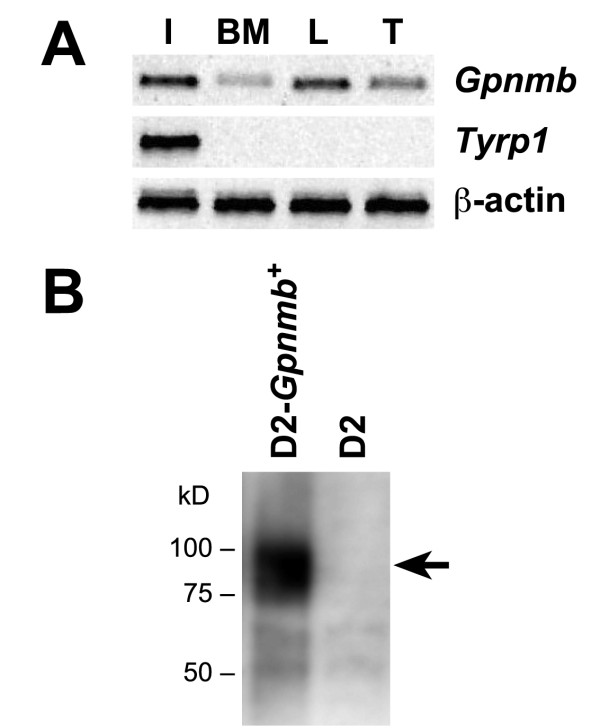
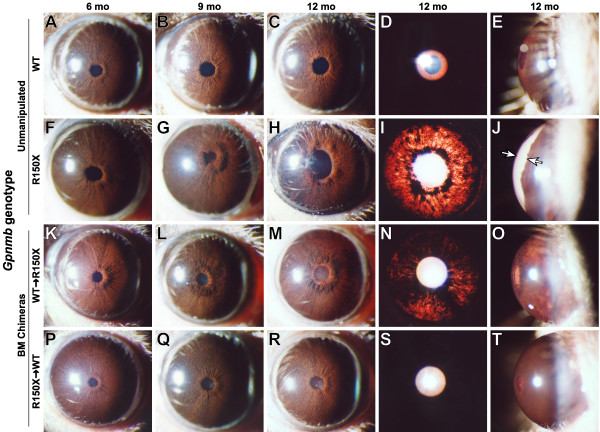
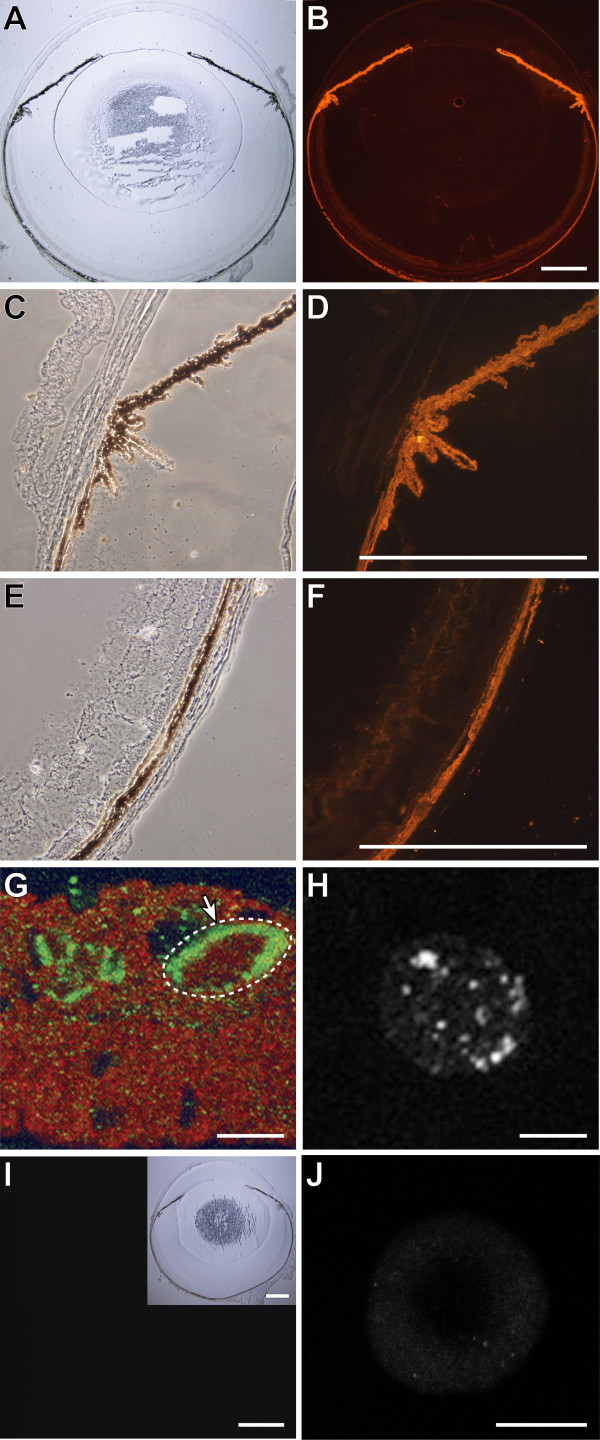
Background: The Gpnmb gene encodes a transmembrane protein whose function(s) remain largely unknown. Here, we assess if a mutant allele of Gpnmb confers susceptibility to glaucoma by altering immune functions. DBA/2J mice have a mutant Gpnmb gene and they develop a form of glaucoma preceded by a pigment dispersing iris disease and abnormalities of the immunosuppressive ocular microenvironment.
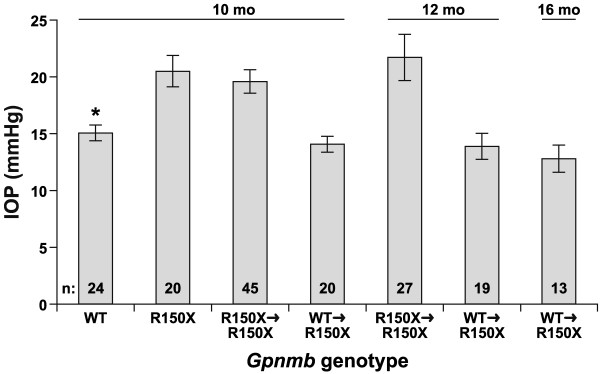
Results: We find that the Gpnmb genotype of bone-marrow derived cell lineages significantly influences the iris disease and the elevation of intraocular pressure. GPNMB localizes to multiple cell types, including pigment producing cells, bone marrow derived F4/80 positive antigen-presenting cells (APCs) of the iris and dendritic cells. We show that APCs of DBA/2J mice fail to induce antigen induced immune deviation (a form of tolerance) when treated with TGFbeta2. This demonstrates that some of the immune abnormalities previously identified in DBA/2J mice result from intrinsic defects in APCs. However, the tested APC defects are not dependent on a mutant Gpnmb gene. Finally, we show that the Gpnmb mediated iris disease does not require elevated IL18 or mature B or T lymphocytes.
Conclusion: These results establish a role for Gpnmb in bone marrow derived lineages. They suggest that affects of Gpnmb on innate immunity influence susceptibility to glaucoma in DBA/2J mice.
Figures

References
-
- Allingham RR, Damji KF, Freedman S, Moroi SE, Shafranov G, Shields MB. Shields' Textbook of Glaucoma. 5. Philadelphia: Lippincott Williams and Wilkins; 2004.
-
- Nickells RW, Jampel HD, Zack DJ. Glaucoma. In: Rimoin DL, Conner MJ, Pyeritz RE, Korf BR, editor. Emery & Rimoins Principles and Practices of Medical Genetics. 4. Vol. 3. Churchill Livingstone; 2002. pp. 3491–3512.
-
- Gordon MO, Beiser JA, Brandt JD, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, Parrish RK, 2nd, Wilson MR, Kass MA. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:714–720. discussion 829–730. - PubMed
-
- Kass MA, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, Parrish RK, 2nd, Wilson MR, Gordon MO. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:701–713. discussion 829–730. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
