

Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries
- PMID: 18403729
- PMCID: PMC2629615
- DOI: 10.1161/CIRCRESAHA.108.172379
Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries
Abstract
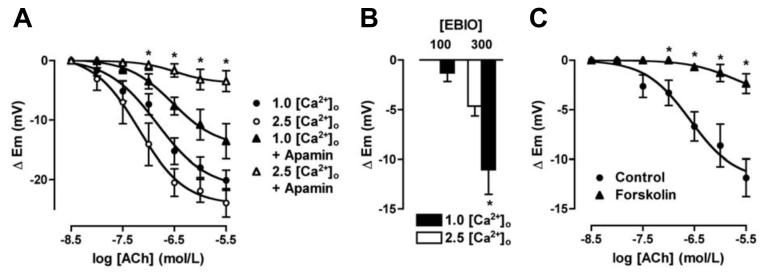
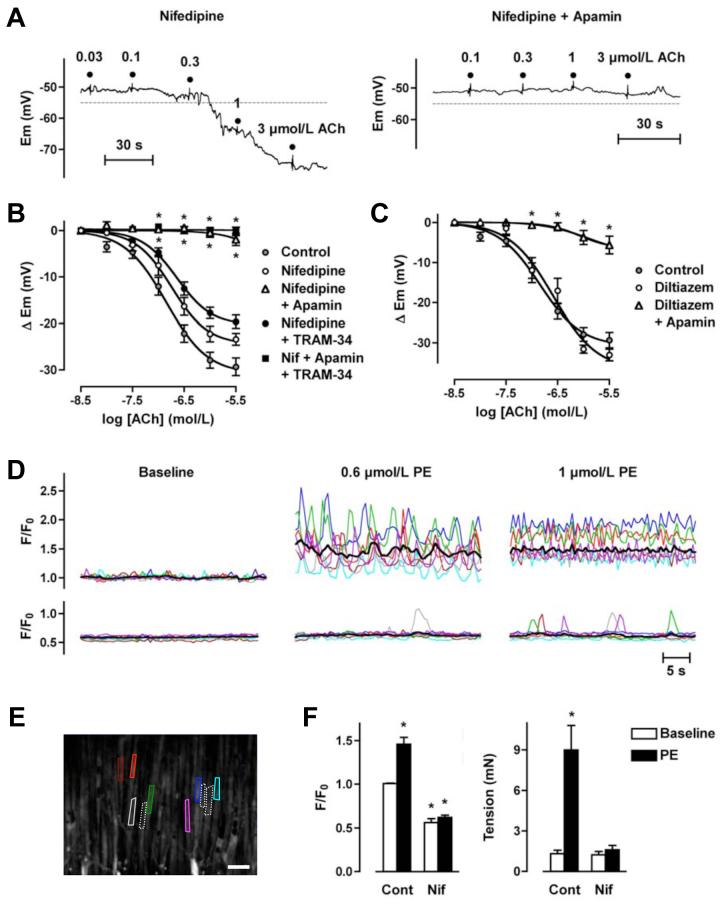
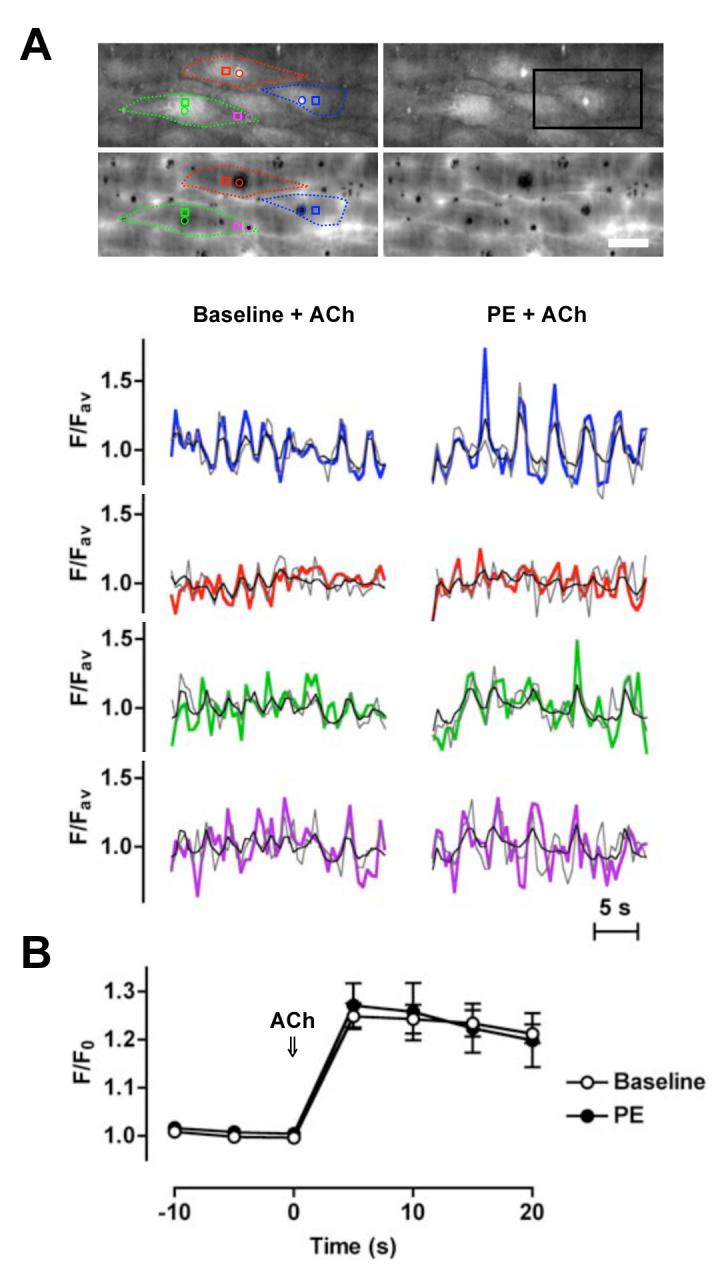
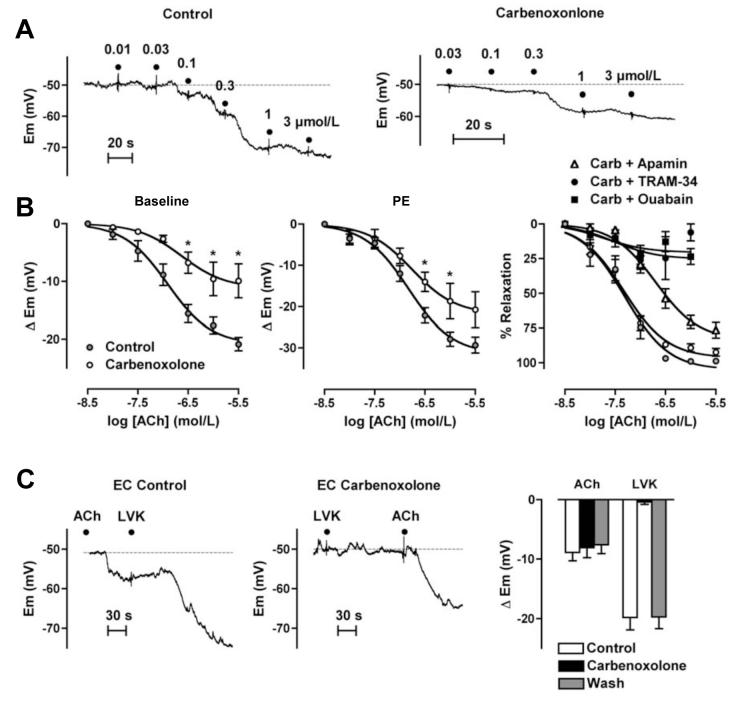
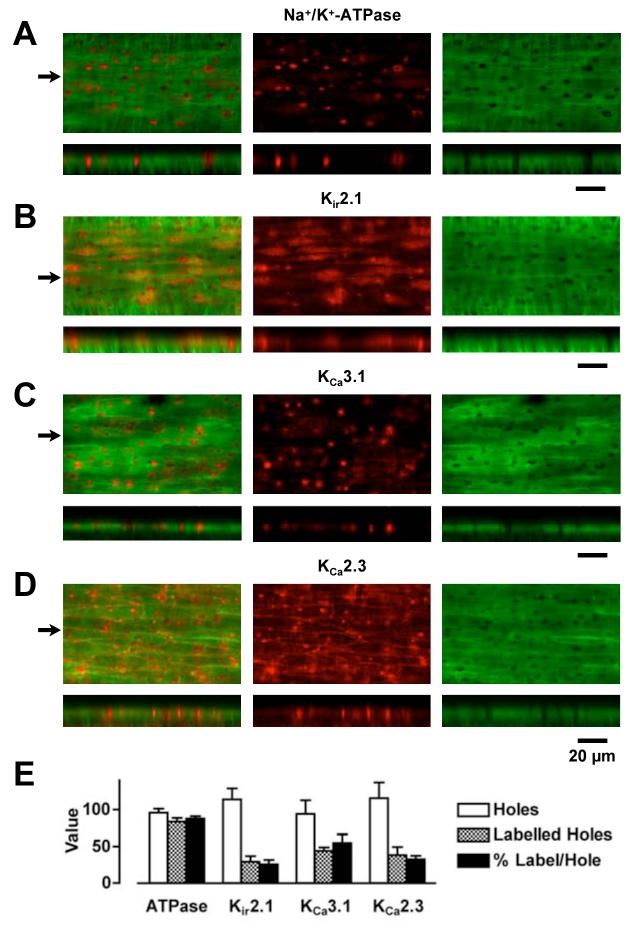
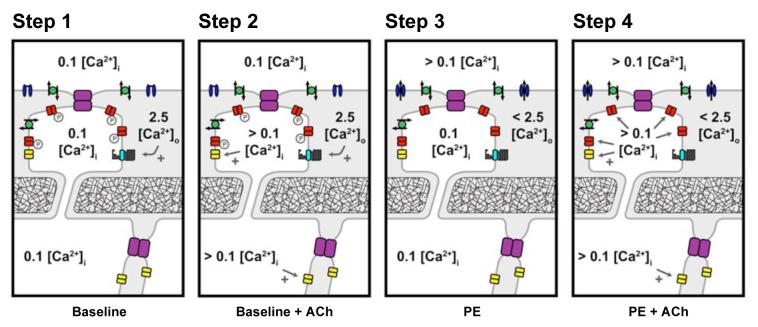
Arterial hyperpolarization to acetylcholine (ACh) reflects coactivation of K(Ca)3.1 (IK(Ca)) channels and K(Ca)2.3 (SK(Ca)) channels in the endothelium that transfers through myoendothelial gap junctions and diffusible factor(s) to affect smooth muscle relaxation (endothelium-derived hyperpolarizing factor [EDHF] response). However, ACh can differentially activate K(Ca)3.1 and K(Ca)2.3 channels, and we investigated the mechanisms responsible in rat mesenteric arteries. K(Ca)3.1 channel input to EDHF hyperpolarization was enhanced by reducing external [Ca(2+)](o) but blocked either with forskolin to activate protein kinase A or by limiting smooth muscle [Ca(2+)](i) increases stimulated by phenylephrine depolarization. Imaging [Ca(2+)](i) within the endothelial cell projections forming myoendothelial gap junctions revealed increases in cytoplasmic [Ca(2+)](i) during endothelial stimulation with ACh that were unaffected by simultaneous increases in muscle [Ca(2+)](i) evoked by phenylephrine. If gap junctions were uncoupled, K(Ca)3.1 channels became the predominant input to EDHF hyperpolarization, and relaxation was inhibited with ouabain, implicating a crucial link through Na(+)/K(+)-ATPase. There was no evidence for an equivalent link through K(Ca)2.3 channels nor between these channels and the putative EDHF pathway involving natriuretic peptide receptor-C. Reconstruction of confocal z-stack images from pressurized arteries revealed K(Ca)2.3 immunostain at endothelial cell borders, including endothelial cell projections, whereas K(Ca)3.1 channels and Na(+)/K(+)-ATPase alpha(2)/alpha(3) subunits were highly concentrated in endothelial cell projections and adjacent to myoendothelial gap junctions. Thus, extracellular [Ca(2+)](o) appears to modify K(Ca)3.1 channel activity through a protein kinase A-dependent mechanism independent of changes in endothelial [Ca(2+)](i). The resulting hyperpolarization links to arterial relaxation largely through Na(+)/K(+)-ATPase, possibly reflecting K(+) acting as an EDHF. In contrast, K(Ca)2.3 hyperpolarization appears mainly to affect relaxation through myoendothelial gap junctions. Overall, these data suggest that K(+) and myoendothelial coupling evoke EDHF-mediated relaxation through distinct, definable pathways.
Figures
Comment in
-
The EDHF story: the plot thickens.Circ Res. 2008 May 23;102(10):1148-50. doi: 10.1161/CIRCRESAHA.108.177279. Circ Res. 2008. PMID: 18497313 No abstract available.
References
-
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. - PubMed
-
- Kohler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, Grgic I, Kampfe D, Si H, Wibawa J, Real R, Borner K, Brakemeier S, Orzechowski HD, Reusch HP, Paul M, Chandy KG, Hoyer J. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 2003;108:1119–1125. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
