

Evaluation of paired-end sequencing strategies for detection of genome rearrangements in cancer
- PMID: 18404202
- PMCID: PMC2278375
- DOI: 10.1371/journal.pcbi.1000051
Evaluation of paired-end sequencing strategies for detection of genome rearrangements in cancer
Abstract
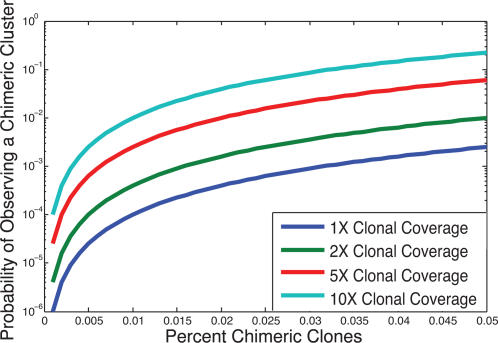
Paired-end sequencing is emerging as a key technique for assessing genome rearrangements and structural variation on a genome-wide scale. This technique is particularly useful for detecting copy-neutral rearrangements, such as inversions and translocations, which are common in cancer and can produce novel fusion genes. We address the question of how much sequencing is required to detect rearrangement breakpoints and to localize them precisely using both theoretical models and simulation. We derive a formula for the probability that a fusion gene exists in a cancer genome given a collection of paired-end sequences from this genome. We use this formula to compute fusion gene probabilities in several breast cancer samples, and we find that we are able to accurately predict fusion genes in these samples with a relatively small number of fragments of large size. We further demonstrate how the ability to detect fusion genes depends on the distribution of gene lengths, and we evaluate how different parameters of a sequencing strategy impact breakpoint detection, breakpoint localization, and fusion gene detection, even in the presence of errors that suggest false rearrangements. These results will be useful in calibrating future cancer sequencing efforts, particularly large-scale studies of many cancer genomes that are enabled by next-generation sequencing technologies.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures








References
-
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. - PubMed
-
- Kurzrock R, Talpaz M. The molecular pathology of chronic myelogenous leukaemia. Br J Haematol. 1991;79:34–37. - PubMed
-
- Druker BJ. STI571 (Gleevec) as a paradigm for cancer therapy. Trends Mol Med. 2002;8:S14–S18. - PubMed
-
- Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet. 2004;36:331–334. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
