

The study of abnormal bone development in the Apert syndrome Fgfr2+/S252W mouse using a 3D hydrogel culture model
- PMID: 18407821
- PMCID: PMC2743143
- DOI: 10.1016/j.bone.2008.02.008
The study of abnormal bone development in the Apert syndrome Fgfr2+/S252W mouse using a 3D hydrogel culture model
Abstract
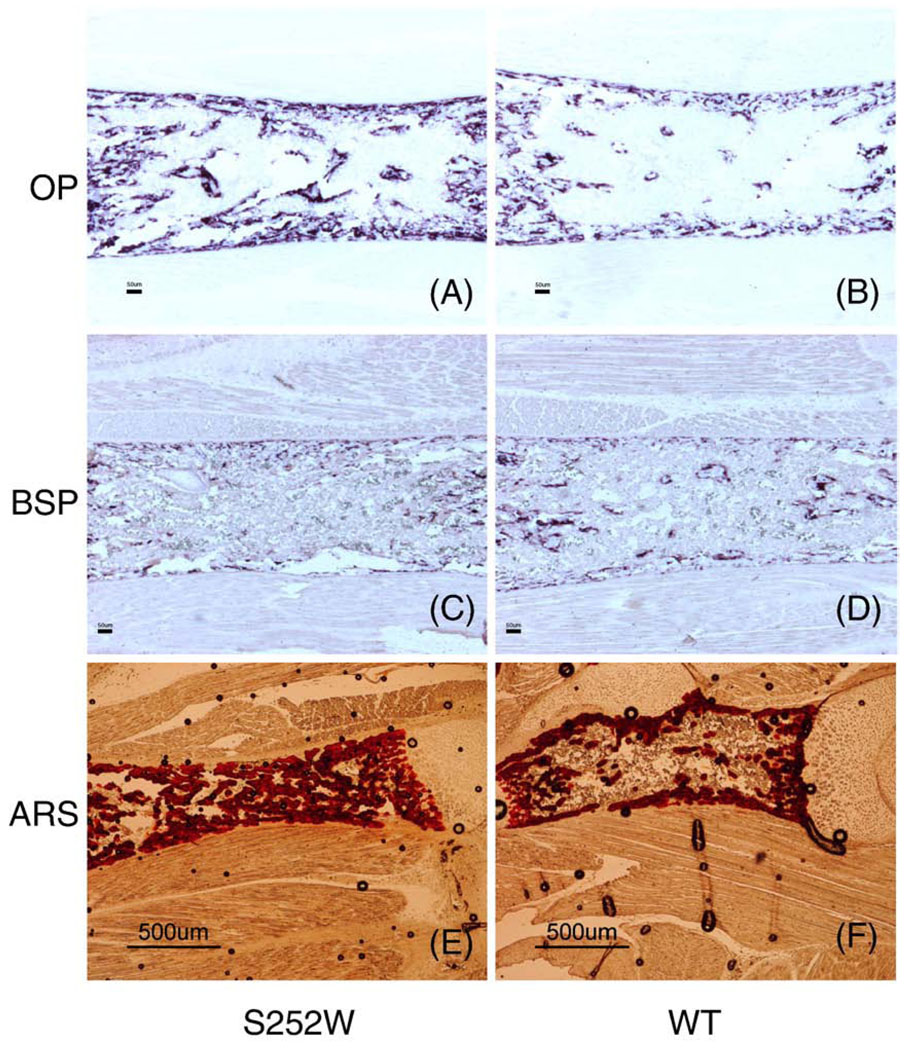
Apert syndrome is caused by mutations in fibroblast growth factor receptor 2 (Fgfr2) and is characterized by craniosynostosis and other skeletal abnormalities. The Apert syndrome Fgfr2+/S252W mouse model exhibits perinatal lethality. A 3D hydrogel culture model, derived from tissue engineering strategies, was used to extend the study of the effect of the Fgfr2+/S252W mutation in differentiating osteoblasts postnatally. We isolated cells from the long bones of Apert Fgfr2+/S252W mice (n=6) and cells from the wild-type sibling mice (n=6) to be used as controls. During monolayer expansion, Fgfr2+/S252W cells demonstrated increased proliferation and ALP activity, as well as altered responses of these cellular functions in the presence of FGF ligands with different binding specificity (FGF2 or FGF10). To better mimic the in vivo disease development scenario, cells were also encapsulated in 3D hydrogels and their phenotype in 3D in vitro culture was compared to that of in vivo tissue specimens. After 4 weeks in 3D culture in osteogenic medium, Fgfr2+/S252W cells expressed 2.8-fold more collagen type I and 3.3-fold more osteocalcin than did wild-type controls (p<0.01). Meanwhile, Fgfr2+/S252W cells showed decreased bone matrix remodeling and expressed 87% less Metalloprotease-13 and 71% less Noggin (p<0.01). The S252W mutation also led to significantly higher production of collagen type I and II in 3D as shown by immunofluorescence staining. In situ hybridization and alizarin red S staining of postnatal day 0 (P0) mouse limb sections demonstrated significantly higher levels of osteopontin expression and mineralization in Fgfr2+/S252W mice. Complementary to in vivo findings, this 3D hydrogel culture system provides an effective in vitro venue to study the pathogenesis of Apert syndrome caused by the analogous mutation in humans.
Conflict of interest statement
All authors have no conflicts of interest.
Figures




Similar articles
-
A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome.J Biol Chem. 2004 Oct 29;279(44):45926-34. doi: 10.1074/jbc.M404824200. Epub 2004 Aug 13. J Biol Chem. 2004. PMID: 15310757
-
The Fgfr2(S252W/+) mutation in mice retards mandible formation and reduces bone mass as in human Apert syndrome.Am J Med Genet A. 2013 May;161A(5):983-92. doi: 10.1002/ajmg.a.35824. Epub 2013 Mar 13. Am J Med Genet A. 2013. PMID: 23495007
-
Aberrantly activated Wnt/β-catenin pathway co-receptors LRP5 and LRP6 regulate osteoblast differentiation in the developing coronal sutures of an Apert syndrome (Fgfr2S252W/+ ) mouse model.Dev Dyn. 2021 Mar;250(3):465-476. doi: 10.1002/dvdy.239. Epub 2020 Sep 9. Dev Dyn. 2021. PMID: 32822074
-
Ovarian dysgerminoma and Apert syndrome.Pediatr Blood Cancer. 2008 Mar;50(3):696-8. doi: 10.1002/pbc.21156. Pediatr Blood Cancer. 2008. PMID: 17243131 Review.
-
[From gene to disease; craniosynostosis syndromes due to FGFR2-mutation].Ned Tijdschr Geneeskd. 2002 Jan 12;146(2):63-6. Ned Tijdschr Geneeskd. 2002. PMID: 11820058 Review. Dutch.
Cited by
-
FGFR2 mutation confers a less drastic gain of function in mesenchymal stem cells than in fibroblasts.Stem Cell Rev Rep. 2012 Sep;8(3):685-95. doi: 10.1007/s12015-011-9327-6. Stem Cell Rev Rep. 2012. PMID: 22048896 Free PMC article.
-
The molecular and cellular basis of Apert syndrome.Intractable Rare Dis Res. 2013 Nov;2(4):115-22. doi: 10.5582/irdr.2013.v2.4.115. Intractable Rare Dis Res. 2013. PMID: 25343114 Free PMC article. Review.
-
PIN1 is a new therapeutic target of craniosynostosis.Hum Mol Genet. 2018 Nov 15;27(22):3827-3839. doi: 10.1093/hmg/ddy252. Hum Mol Genet. 2018. PMID: 30007339 Free PMC article.
-
Visualization of the Cartilage and Bone Elements in the Craniofacial Structures by Alcian Blue and Alizarin Red Staining.Methods Mol Biol. 2022;2403:43-50. doi: 10.1007/978-1-0716-1847-9_4. Methods Mol Biol. 2022. PMID: 34913115
-
Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology.Dev Biol. 2009 Apr 15;328(2):273-84. doi: 10.1016/j.ydbio.2009.01.026. Epub 2009 Jan 29. Dev Biol. 2009. PMID: 19389359 Free PMC article.
References
-
- Chen L, Deng CX. Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci. 2005;10:1961–1976. - PubMed
-
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. - PubMed
-
- Cohen MM, Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, Erickson JD, Roeper P, Martinez-Frias ML. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992;42:655–659. - PubMed
-
- Cohen MM, Jr, Kreiborg S. New indirect method for estimating the birth prevalence of the Apert syndrome. Int J Oral Maxillofac Surg. 1992;21:107–109. - PubMed
-
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–172. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
