

Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation
- PMID: 18411341
- PMCID: PMC2373837
- DOI: 10.1084/jem.20072528
Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation
Abstract
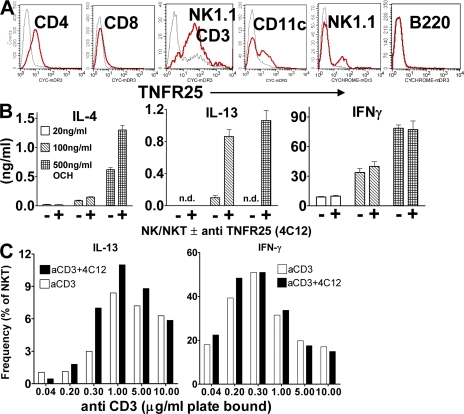
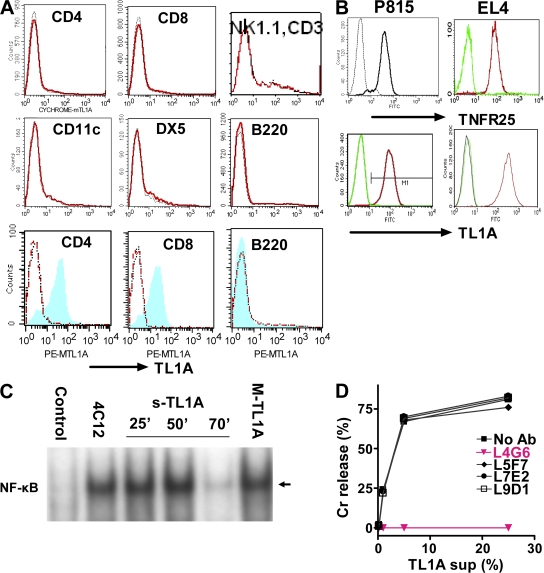
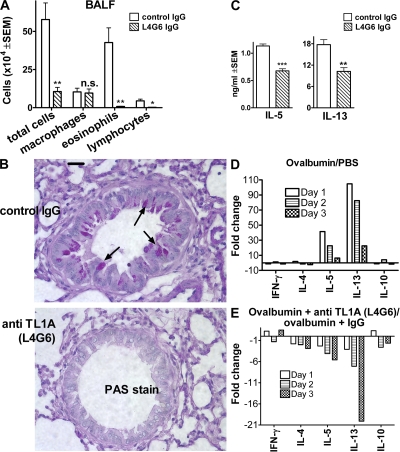
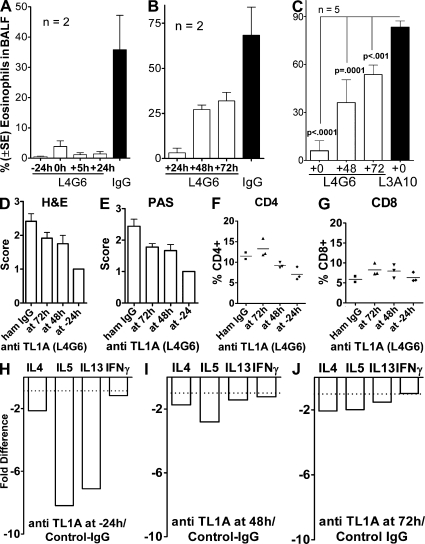
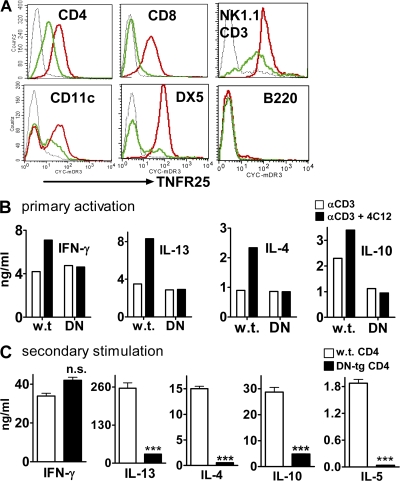
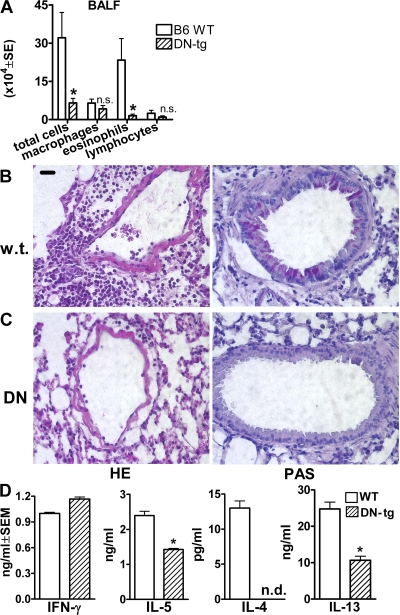
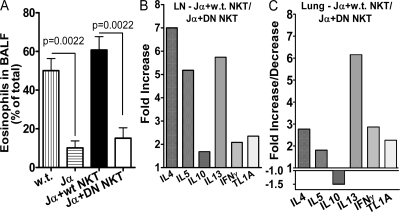
We identify the tumor necrosis factor receptor superfamily 25 (TNFRSF25)/TNFSF15 pair as critical trigger for allergic lung inflammation, which is a cardinal feature of asthma. TNFRSF25 (TNFR25) signals are required to exert T helper cell 2 (Th2) effector function in Th2-polarized CD4 cells and co-stimulate interleukin (IL)-13 production by glycosphingolipid-activated NKT cells. In vivo, antibody blockade of TNFSF15 (TL1A), which is the ligand for TNFR25, inhibits lung inflammation and production of Th2 cytokines such as IL-13, even when administered days after airway antigen exposure. Similarly, blockade of TNFR25 by a dominant-negative (DN) transgene, DN TNFR25, confers resistance to lung inflammation in mice. Allergic lung inflammation-resistant, NKT-deficient mice become susceptible upon adoptive transfer of wild-type NKT cells, but not after transfer of DN TNFR25 transgenic NKT cells. The TNFR25/TL1A pair appears to provide an early signal for Th2 cytokine production in the lung, and therefore may be a drug target in attempts to attenuate lung inflammation in asthmatics.
Figures
References
-
- Chinnaiyan, A.M., K. O'Rourke, G.L. Yu, R.H. Lyons, M. Garg, D.R. Duan, L. Xing, R. Gentz, J. Ni, and V.M. Dixit. 1996. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 274:990–992. - PubMed
-
- Kitson, J., T. Raven, Y.P. Jiang, D.V. Goeddel, K.M. Giles, K.T. Pun, C.J. Grinham, R. Brown, and S.N. Farrow. 1996. A death-domain-containing receptor that mediates apoptosis. Nature. 384:372–375. - PubMed
-
- Bodmer, J.L., K. Burns, P. Schneider, K. Hofmann, V. Steiner, M. Thome, T. Bornand, M. Hahne, M. Schroter, K. Becker, et al. 1997. TRAMP, a novel apoptosis-mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas(Apo-1/CD95). Immunity. 6:79–88. - PubMed
-
- Marsters, S.A., J.P. Sheridan, C.J. Donahue, R.M. Pitti, C.L. Gray, A.D. Goddard, K.D. Bauer, and A. Ashkenazi. 1996. Apo-3, a new member of the tumor necrosis factor receptor family, contains a death domain and activates apoptosis and NF-kappa B. Curr. Biol. 6:1669–1676. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
