

Accelerating QM/MM free energy calculations: representing the surroundings by an updated mean charge distribution
- PMID: 18412414
- PMCID: PMC2651397
- DOI: 10.1021/jp711496y
Accelerating QM/MM free energy calculations: representing the surroundings by an updated mean charge distribution
Abstract
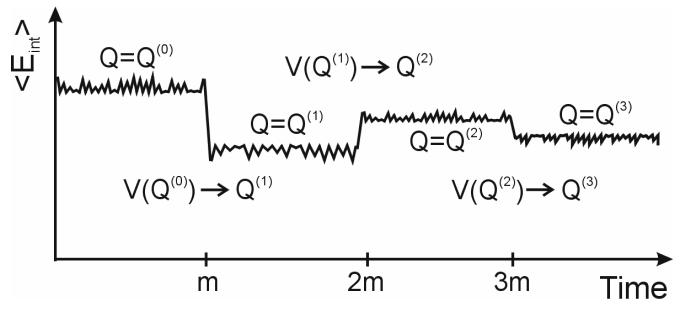
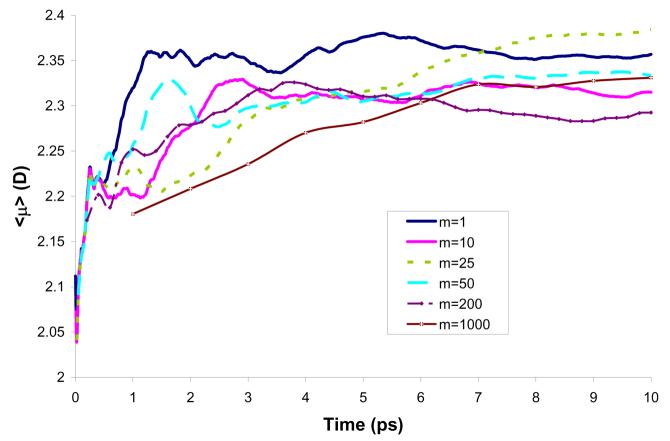
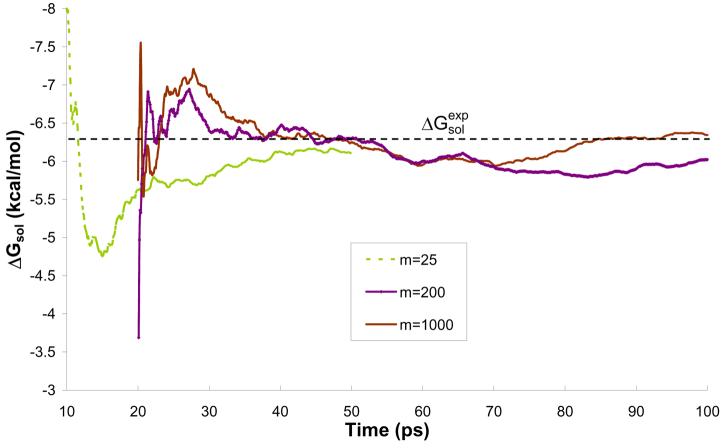
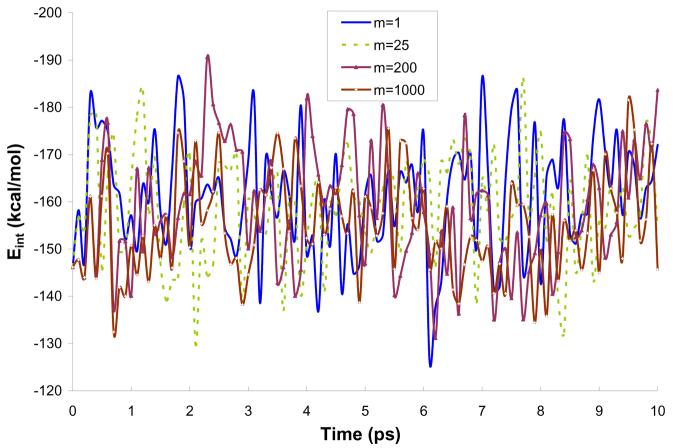
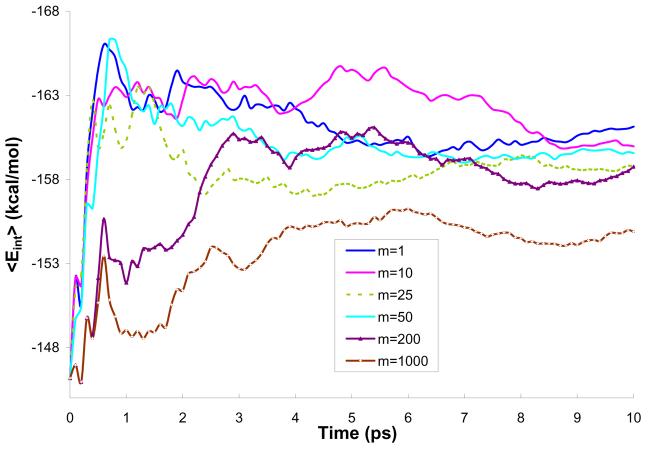
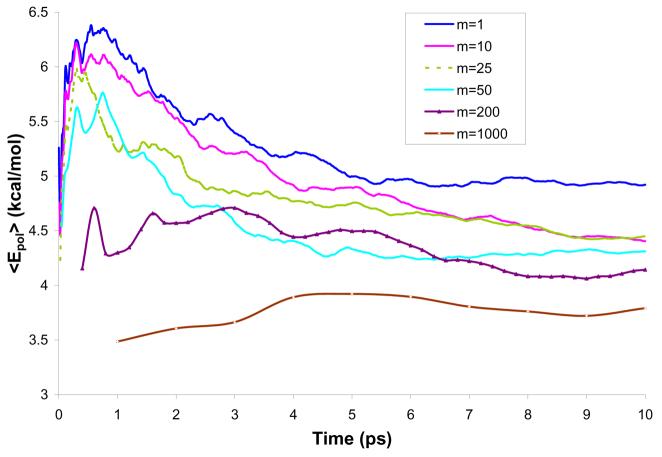
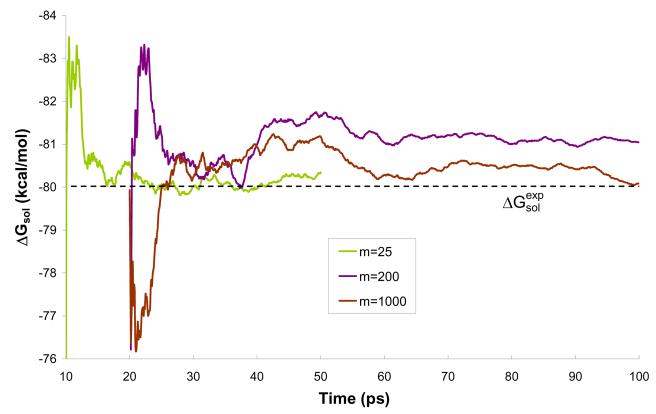
Reliable studies of enzymatic reactions by combined quantum mechanical/molecular mechanics (QM(ai)/MM) approaches with an ab initio description of the quantum region presents a major challenge to computational chemists. The main problem is the need for very large computer time to evaluate the QM energy, which in turn makes it extremely challenging to perform proper configurational sampling. One of the most obvious options for accelerating QM/MM simulations is the use of an average solvent potential. In fact, the idea of using an average solvent potential is rather obvious and has implicitly been used in Langevin dipole/QM calculations. However, in the case of explicit solvent models the practical implementations are more challenging, and the accuracy of the averaging approach has not been validated. The present study introduces the average effect of the fluctuating solvent charges by using equivalent charge distributions, which are updated every m steps. Several models are evaluated in terms of the resulting accuracy and efficiency. The most effective model divides the system into an inner region with N explicit solvent atoms and an external region with two effective charges. Different models are considered in terms of the division of the solvent system and the update frequency. Another key element of our approach is the use of the free energy perturbation (FEP) and/or linear response approximation treatments that guarantees the evaluation of the rigorous solvation free energy. Special attention is paid to the convergence of the calculated solvation free energies and the corresponding solute polarization. The performance of the method is examined by evaluating the solvation of a water molecule and a formate ion in water and also the dipole moment of water in water solution. Remarkably, it is found that different averaging procedures eventually converge to the same value but some protocols provide optimal ways of obtaining the final QM(ai)/MM converged results. The current method can provide computational time saving of 1000 for properly converging simulations relative to calculations that evaluate the QM(ai)/MM energy every time step. A specialized version of our approach that starts with a classical FEP charging and then evaluates the free energy of moving from the classical potential to the QM/MM potential appears to be particularly effective. This approach should provide a very powerful tool for QM(ai)/MM evaluation of solvation free energies in aqueous solutions and proteins.
Figures
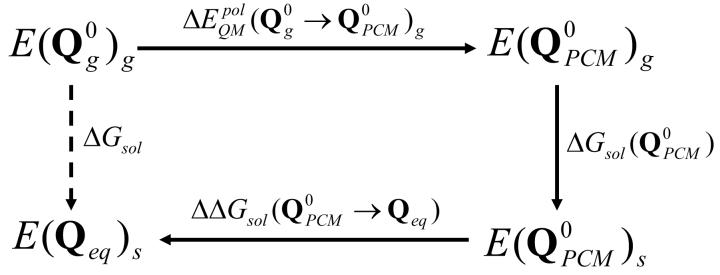
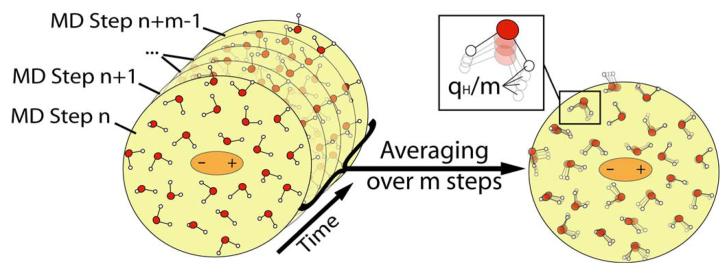
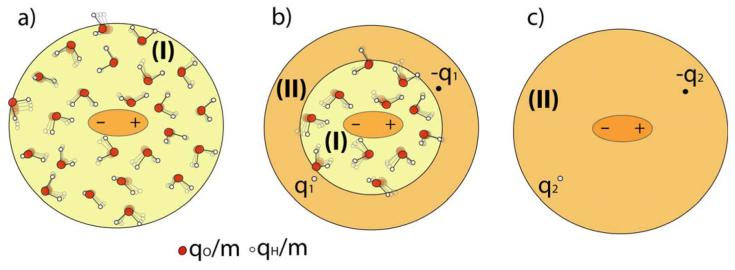
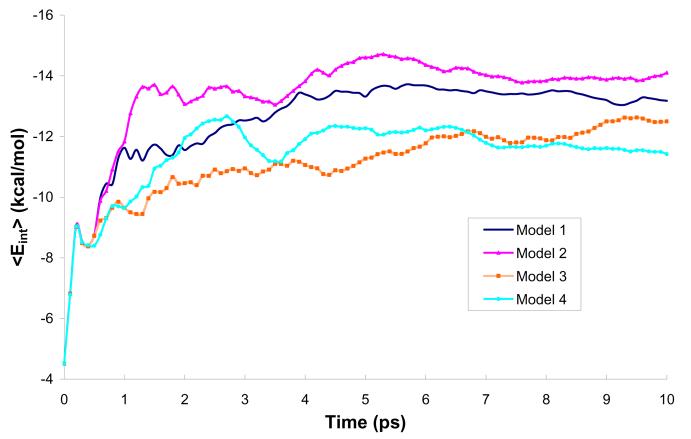
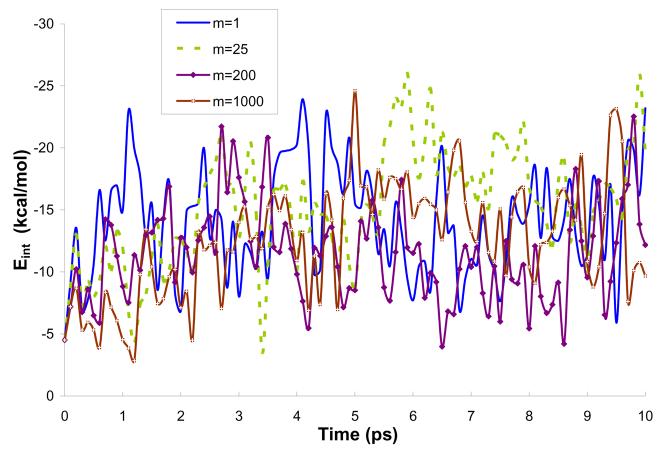
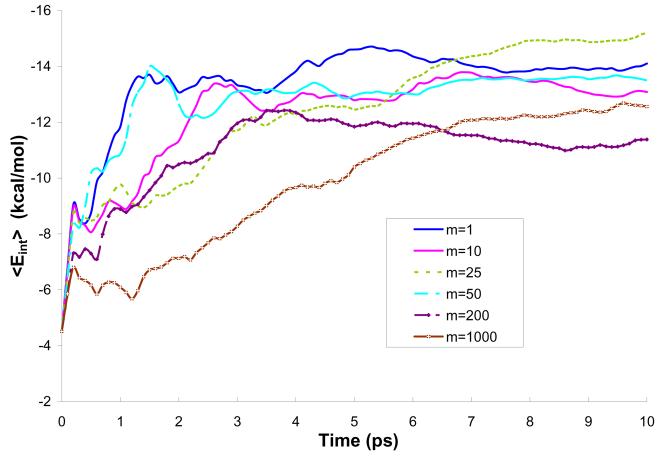
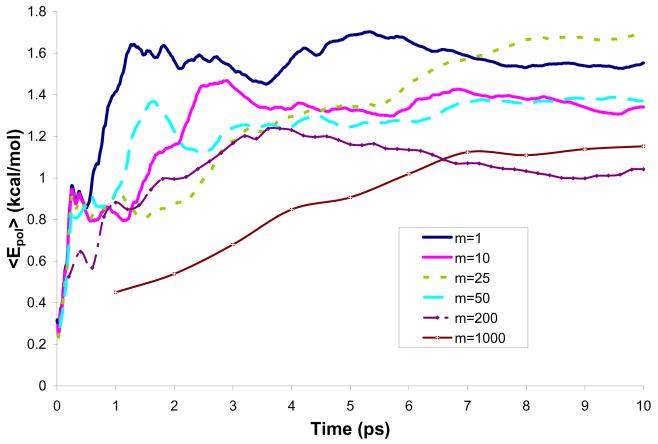
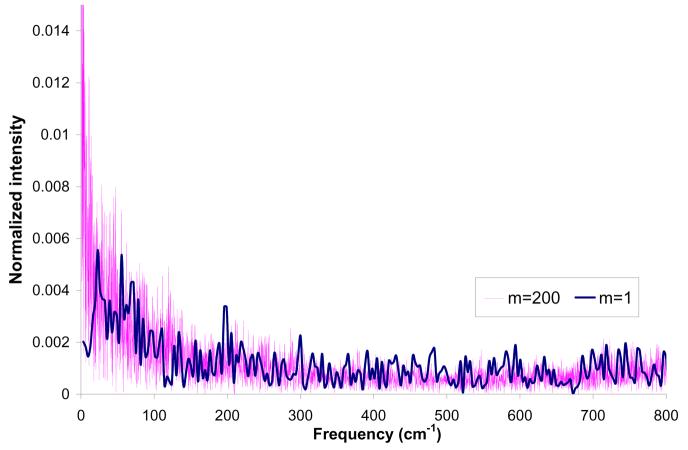
References
-
- Warshel A, Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976;103:227–249. - PubMed
-
- Shurki A, Warshel A. Structure/Function Correlations of Protreins using MM, QM/MM and Related Approaches; Methods, Concepts, Pitfalls and Current Progress. Advances in Protein Chemistry. 2003;66:249–312. - PubMed
-
- Gao J. Hybrid quantum and molecular mechanical simulations: an alternative avenue to solvent effects in organic chemistry. Acc. Chem. Res. 1996;29:298–305.
-
- Bakowies D, Thiel W. Hybrid Models for Combined Quantum Mechanical and Molecular Approaches. J. Phys. Chem. 1996;100:10580–10594.
-
- Field MJ, Bash PA, Karplus M. A Combined Quantum Mechanical and Molecular Mechanical Potential for Molecular Dynamics Simulations. J. Comp. Chem. 1990;11:700–733.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
