

Lack of spartin protein in Troyer syndrome: a loss-of-function disease mechanism?
- PMID: 18413476
- PMCID: PMC5580255
- DOI: 10.1001/archneur.65.4.520
Lack of spartin protein in Troyer syndrome: a loss-of-function disease mechanism?
Abstract
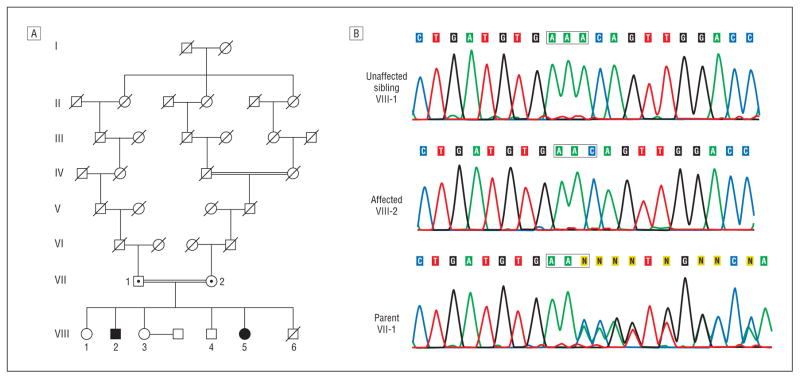
Background: Hereditary spastic paraplegias (SPG1-SPG33) are characterized by progressive spastic weakness of the lower limbs. A nucleotide deletion (1110delA) in the (SPG20; OMIM 275900) spartin gene is the origin of autosomal recessive Troyer syndrome. This mutation is predicted to cause premature termination of the spartin protein. However, it remains unknown whether this truncated spartin protein is absent or is present and partially functional in patients.
Objective: To determine whether the truncated spartin protein is present or absent in cells derived from patients with Troyer syndrome.
Design: Case report.
Setting: Academic research.
Patients: We describe a new family with Troyer syndrome due to the 1110delA mutation.
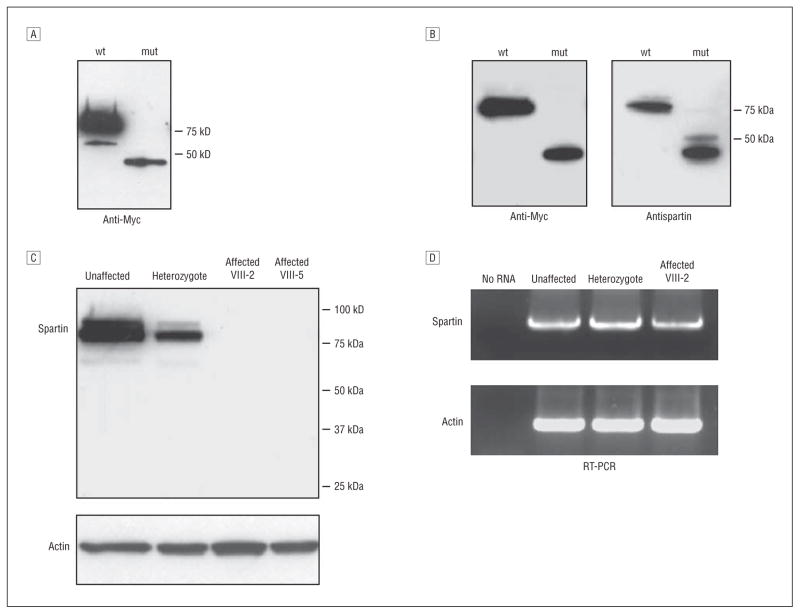
Main outcome measures: We cultured primary fibroblasts and generated lymphoblasts from affected individuals, carriers, and control subjects and subjected these cells to immunoblot analyses.
Results: Spartin protein is undetectable in several cell lines derived from patients with Troyer syndrome.
Conclusions: Our data suggest that Troyer syndrome results from complete loss of spartin protein rather than from the predicted partly functional fragment. This may reflect increased protein degradation or impaired translation.
Figures
References
-
- Harding AE. Hereditary spastic paraplegias. Semin Neurol. 1993;13(4):333–336. - PubMed
-
- Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6(1):65–76. - PubMed
-
- Soderblom C, Blackstone C. Traffic accidents: molecular genetic insights into the pathogenesis of the hereditary spastic paraplegias. Pharmacol Ther. 2006;109(1–2):42–56. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
