

Activation of the lifespan regulator p66Shc through reversible disulfide bond formation
- PMID: 18413607
- PMCID: PMC2311372
- DOI: 10.1073/pnas.0800691105
Activation of the lifespan regulator p66Shc through reversible disulfide bond formation
Abstract
Cell fate and organismal lifespan are controlled by a complex signaling network whose dysfunction can cause a variety of aging-related diseases. An important protection against these failures is cellular apoptosis, which can be induced by p66(Shc) in response to cellular stress. The precise mechanisms of p66(Shc) action and regulation and the function of the p66(Shc)-specific N terminus remain to be identified. Here, we show that the p66(Shc) N terminus forms a redox module responsible for apoptosis initiation, and that this module can be activated through reversible tetramerization by forming two disulfide bonds. Glutathione and thioredoxins can reduce and inactivate p66(Shc), resulting in a thiol-based redox sensor system that initiates apoptosis once cellular protection systems cannot cope anymore with cellular stress.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
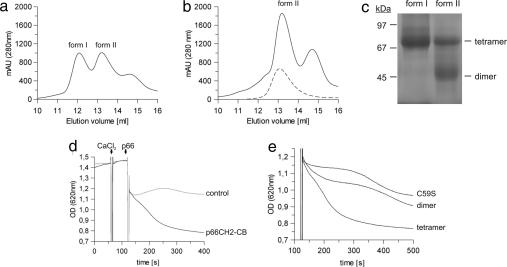
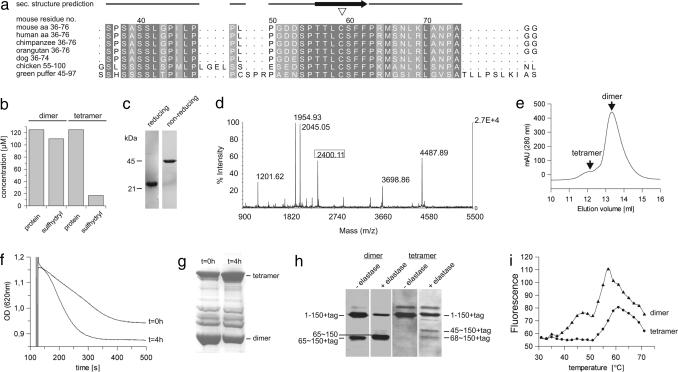
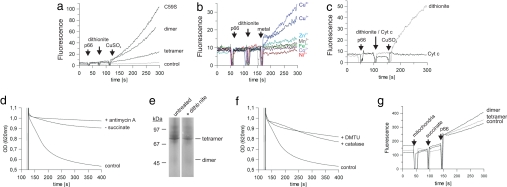
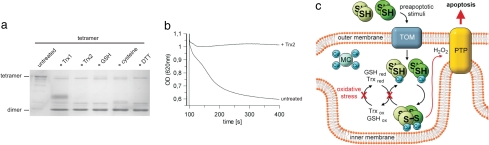
References
-
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. - PubMed
-
- Lee HC, Wei YH. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med. 2007;232:592–606. - PubMed
-
- Hekimi S. How genetic analysis tests theories of animal aging. Nat Genet. 2006;38:985–991. - PubMed
-
- Migliaccio E, et al. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
