

Cardiogenic small molecules that enhance myocardial repair by stem cells
- PMID: 18420817
- PMCID: PMC2329693
- DOI: 10.1073/pnas.0711507105
Cardiogenic small molecules that enhance myocardial repair by stem cells
Abstract
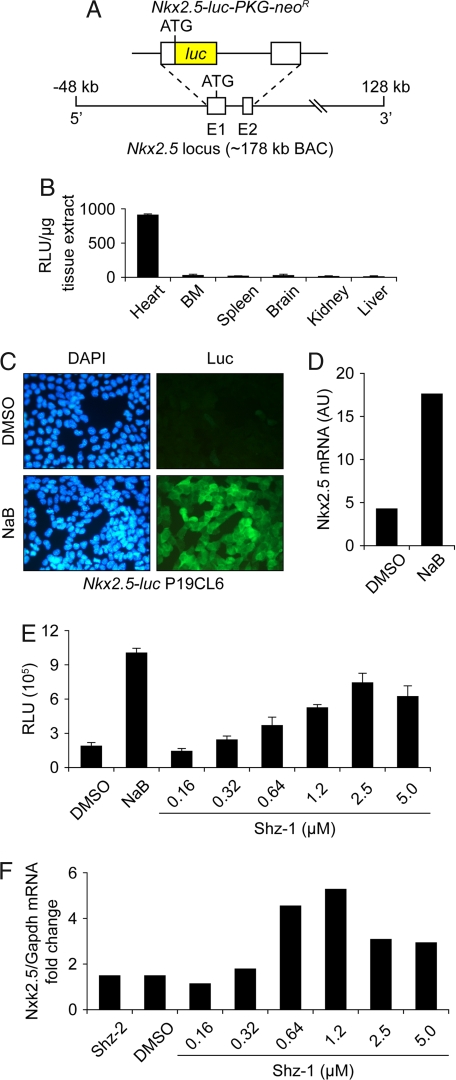
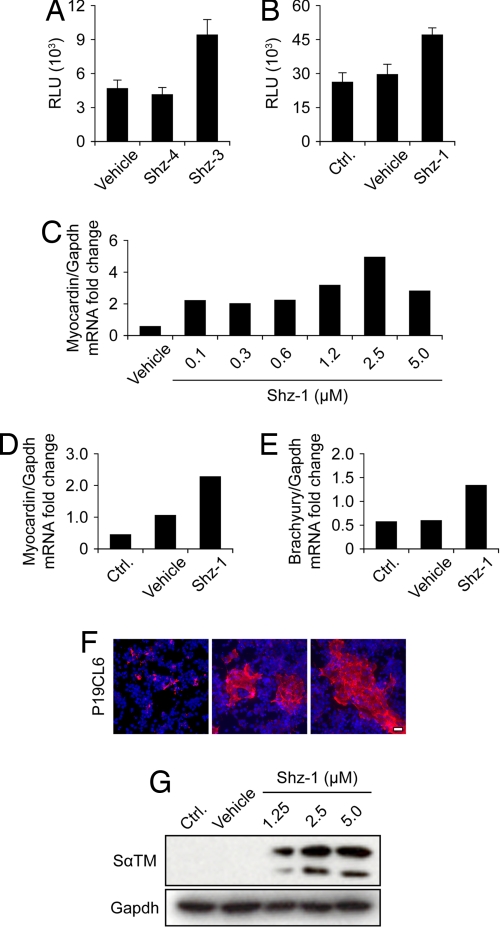
The clinical success of stem cell therapy for myocardial repair hinges on a better understanding of cardiac fate mechanisms. We have identified small molecules involved in cardiac fate by screening a chemical library for activators of the signature gene Nkx2.5, using a luciferase knockin bacterial artificial chromosome (BAC) in mouse P19CL6 pluripotent stem cells. We describe a family of sulfonyl-hydrazone (Shz) small molecules that can trigger cardiac mRNA and protein expression in a variety of embryonic and adult stem/progenitor cells, including human mobilized peripheral blood mononuclear cells (M-PBMCs). Small-molecule-enhanced M-PBMCs engrafted into the rat heart in proximity to an experimental injury improved cardiac function better than control cells. Recovery of cardiac function correlated with persistence of viable human cells, expressing human-specific cardiac mRNAs and proteins. Shz small molecules are promising starting points for drugs to promote myocardial repair/regeneration by activating cardiac differentiation in M-PBMCs.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Wojakowski W, Kucia M, Kazmierski M, Ratajczak MZ, Tendera M. Circulating stem/progenitor cells in stable ischemic heart disease and acute coronary syndromes—relevant reparatory mechanism? Heart. 2008;94:27–33. - PubMed
-
- Rosenzweig A. Cardiac cell therapy—mixed results from mixed cells. New Engl J Med. 2006;355:1274–1277. - PubMed
-
- Assmus B, et al. Transcoronary transplantation of progenitor cells after myocardial infarction. New Engl J Med. 2006;355:1222–1232. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
