

Functional maps of protein complexes from quantitative genetic interaction data
- PMID: 18421374
- PMCID: PMC2289880
- DOI: 10.1371/journal.pcbi.1000065
Functional maps of protein complexes from quantitative genetic interaction data
Abstract
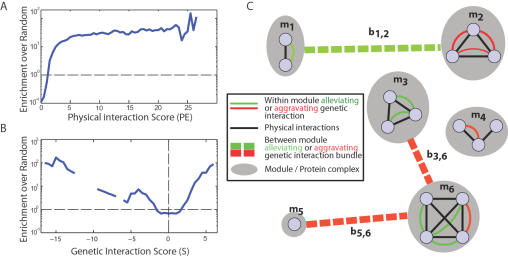
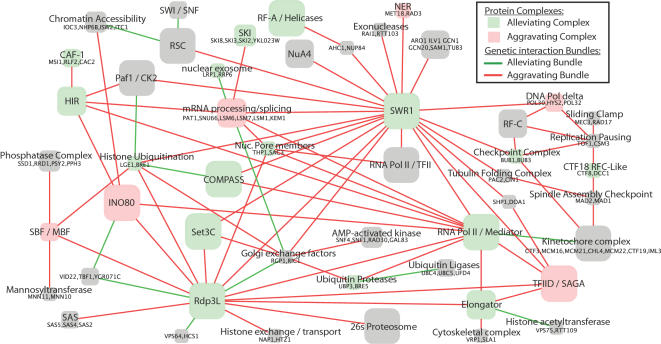
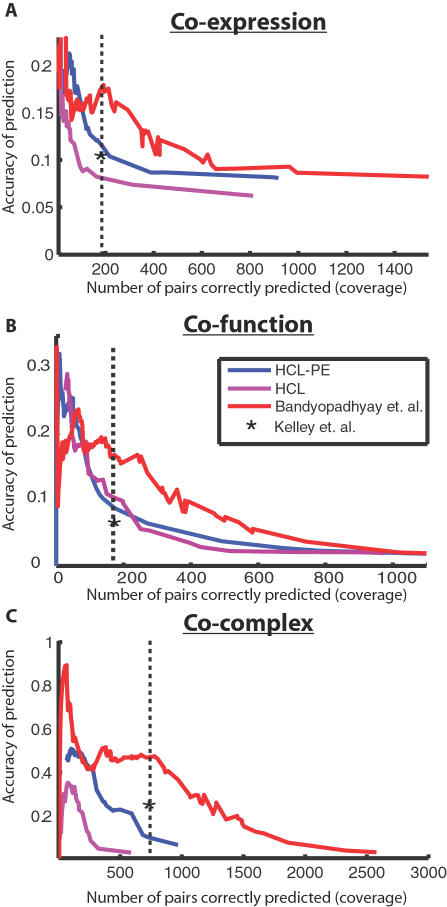
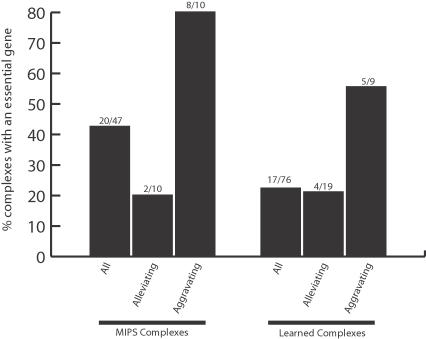
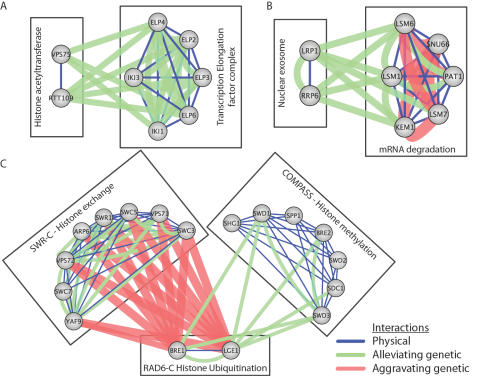
Recently, a number of advanced screening technologies have allowed for the comprehensive quantification of aggravating and alleviating genetic interactions among gene pairs. In parallel, TAP-MS studies (tandem affinity purification followed by mass spectroscopy) have been successful at identifying physical protein interactions that can indicate proteins participating in the same molecular complex. Here, we propose a method for the joint learning of protein complexes and their functional relationships by integration of quantitative genetic interactions and TAP-MS data. Using 3 independent benchmark datasets, we demonstrate that this method is >50% more accurate at identifying functionally related protein pairs than previous approaches. Application to genes involved in yeast chromosome organization identifies a functional map of 91 multimeric complexes, a number of which are novel or have been substantially expanded by addition of new subunits. Interestingly, we find that complexes that are enriched for aggravating genetic interactions (i.e., synthetic lethality) are more likely to contain essential genes, linking each of these interactions to an underlying mechanism. These results demonstrate the importance of both large-scale genetic and physical interaction data in mapping pathway architecture and function.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Hereford LM, Hartwell LH. Sequential gene function in the initiation of Saccharomyces cerevisiae DNA synthesis. J Mol Biol. 1974;84:445–461. - PubMed
-
- Ooi SL, Shoemaker DD, Boeke JD. DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat Genet. 2003;35:277–286. - PubMed
-
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
