

Using galaxy to perform large-scale interactive data analyses
- PMID: 18428782
- PMCID: PMC3418382
- DOI: 10.1002/0471250953.bi1005s19
Using galaxy to perform large-scale interactive data analyses
Abstract
While most experimental biologists know where to download genomic data, few have a concrete plan on how to analyze it. This situation can be corrected by: (1) providing unified portals serving genomic data and (2) building Web applications to allow flexible retrieval and on-the-fly analyses of the data. Powerful resources, such as the UCSC Genome Browser already address the first issue. The second issue, however, remains open. For example, how to find human protein-coding exons with the highest density of single nucleotide polymorphisms (SNPs) and extract orthologous sequences from all sequenced mammals? Indeed, one can access all relevant data from the UCSC Genome Browser. But once the data is downloaded how would one deal with millions of SNPs and gigabytes of alignments? Galaxy (http://g2.bx.psu.edu) is designed specifically for that purpose. It amplifies the strengths of existing resources (such as UCSC Genome Browser) by allowing the user to access and, most importantly, analyze data within a single interface in an unprecedented number of ways.
Copyright 2007 by John Wiley & Sons, Inc.
Figures




































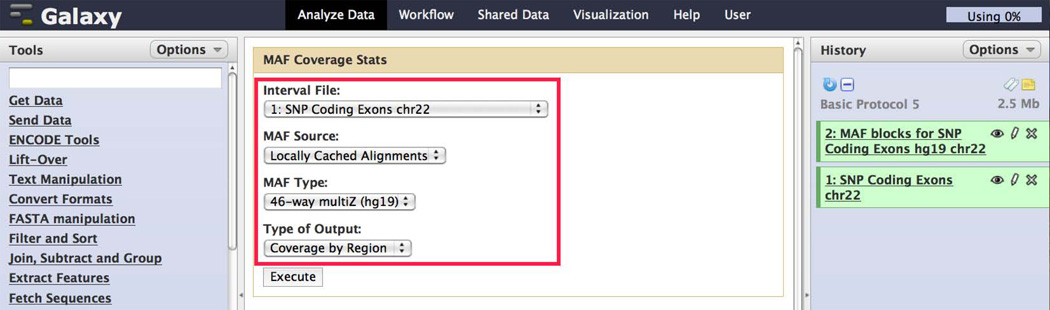












References
-
- Birney E, Andrews D, Bevan P, Caccamo M, Cameron G, Chen Y, Clarke L, Coates G, Cox T, Cuff J, Curwen V, Cutts T, Down T, Durbin R, Eyras E, Fernandez-Suarez XM, Gane P, Gibbins B, Gilbert J, Hammond M, Hotz H, Iyer V, Kahari A, Jekosch K, Kasprzyk A, Keefe D, Keenan S, Lehvaslaiho H, McVicker G, Melsopp C, Meidl P, Mongin E, Pettett R, Potter S, Proctor G, Rae M, Searle S, Slater G, Smedley D, Smith J, Spooner W, Stabenau A, Stalker J, Storey R, Ureta-Vidal A, Woodwark C, Clamp M, Hubbard T. Ensembl 2004. Nucl. Acids Res. 2004;32:D468–D470. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
