

Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation
- PMID: 18431490
- PMCID: PMC2329593
- DOI: 10.1371/journal.pone.0002009
Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation
Abstract
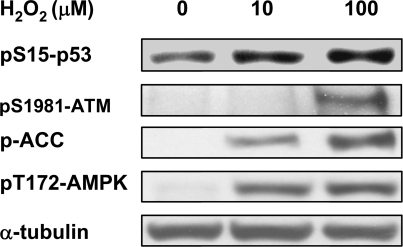
Background: DNA damage such as double-stranded DNA breaks (DSBs) has been reported to stimulate mitochondrial biogenesis. However, the underlying mechanism is poorly understood. The major player in response to DSBs is ATM (ataxia telangiectasia mutated). Upon sensing DSBs, ATM is activated through autophosphorylation and phosphorylates a number of substrates for DNA repair, cell cycle regulation and apoptosis. ATM has been reported to phosphorylate the alpha subunit of AMP-activated protein kinase (AMPK), which senses AMP/ATP ratio in cells, and can be activated by upstream kinases. Here we provide evidence for a novel role of ATM in mitochondrial biogenesis through AMPK activation in response to etoposide-induced DNA damage.
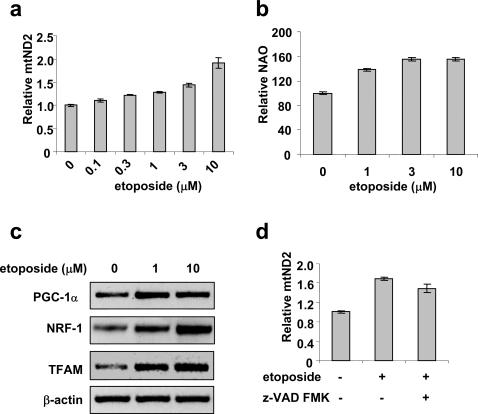
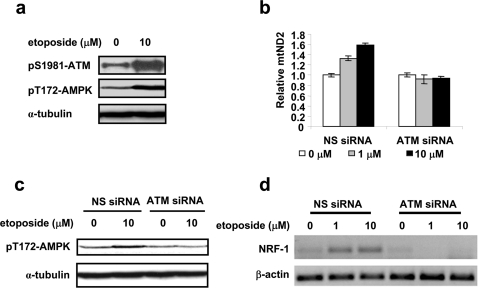
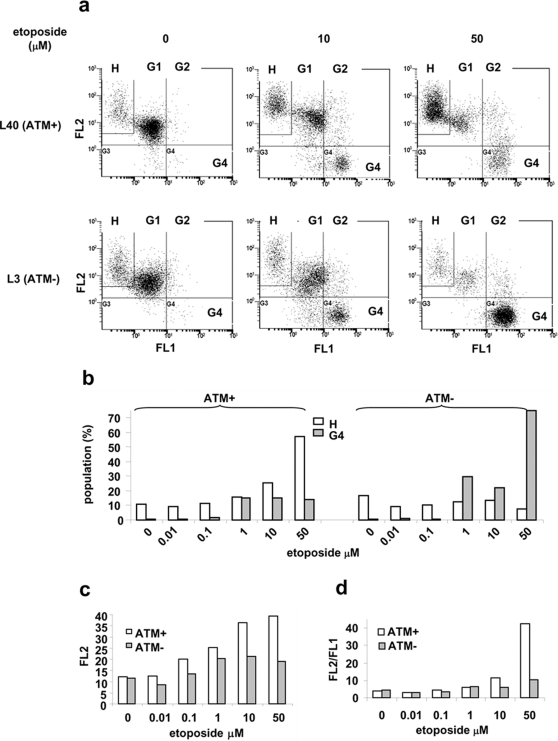
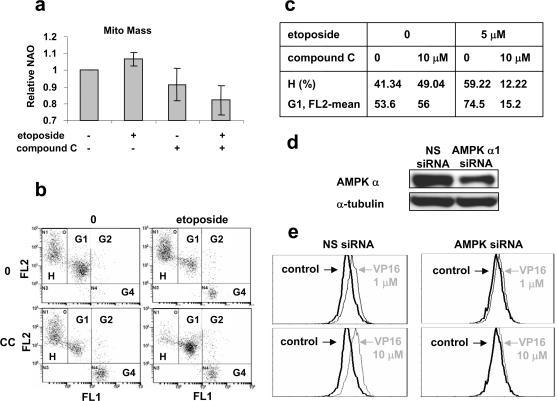
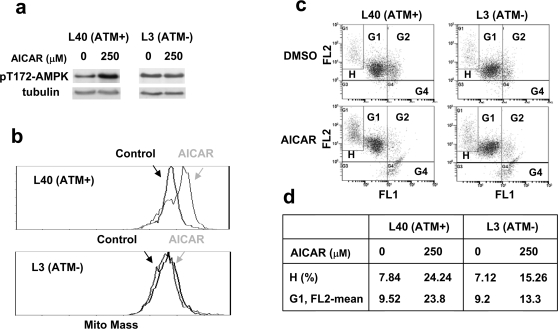
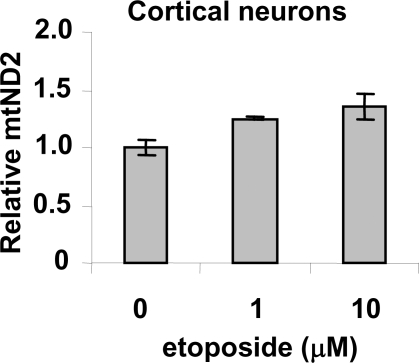
Methodology/principal findings: Three pairs of human ATM+ and ATM- cells were employed. Cells treated with etoposide exhibited an ATM-dependent increase in mitochondrial mass as measured by 10-N-Nonyl-Acridine Orange and MitoTracker Green FM staining, as well as an increase in mitochondrial DNA content. In addition, the expression of several known mitochondrial biogenesis regulators such as the major mitochondrial transcription factor NRF-1, PGC-1alpha and TFAM was also elevated in response to etoposide treatment as monitored by RT-PCR. Three pieces of evidence suggest that etoposide-induced mitochondrial biogenesis is due to ATM-dependent activation of AMPK. First, etoposide induced ATM-dependent phosphorylation of AMPK alpha subunit at Thr172, indicative of AMPK activation. Second, inhibition of AMPK blocked etoposide-induced mitochondrial biogenesis. Third, activation of AMPK by AICAR (an AMP analogue) stimulated mitochondrial biogenesis in an ATM-dependent manner, suggesting that ATM may be an upstream kinase of AMPK in the mitochondrial biogenesis pathway.
Conclusions/significance: These results suggest that activation of ATM by etoposide can lead to mitochondrial biogenesis through AMPK activation. We propose that ATM-dependent mitochondrial biogenesis may play a role in DNA damage response and ROS regulation, and that defect in ATM-dependent mitochondrial biogenesis could contribute to the manifestations of A-T disease.
Conflict of interest statement
Figures
References
-
- Zeviani M, Carelli V. Mitochondrial disorders. Curr Opin Neurol. 2003;16:585–594. - PubMed
-
- Sadun AA, Carelli V. Mitochondrial function and dysfunction within the optic nerve. Arch Ophthalmol. 2003;121:1342–1343. - PubMed
-
- Zeviani M, Spinazzola A, Carelli V. Nuclear genes in mitochondrial disorders. Curr Opin Genet Dev. 2003;13:262–270. - PubMed
-
- Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
