

Identification of kinetin riboside as a repressor of CCND1 and CCND2 with preclinical antimyeloma activity
- PMID: 18431519
- PMCID: PMC2323188
- DOI: 10.1172/JCI34149
Identification of kinetin riboside as a repressor of CCND1 and CCND2 with preclinical antimyeloma activity
Abstract
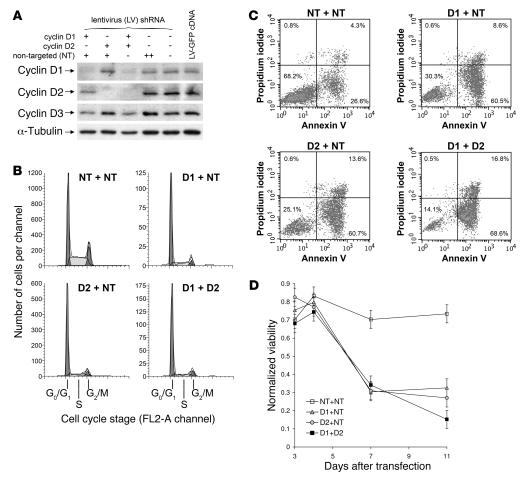
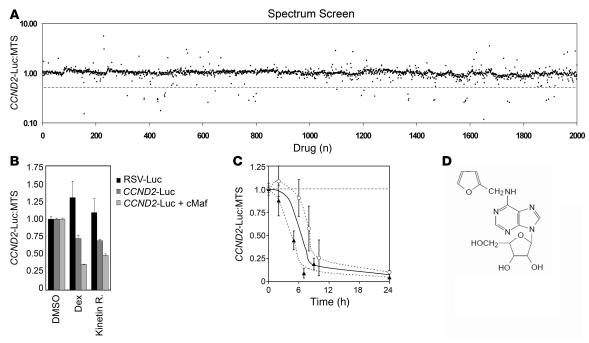
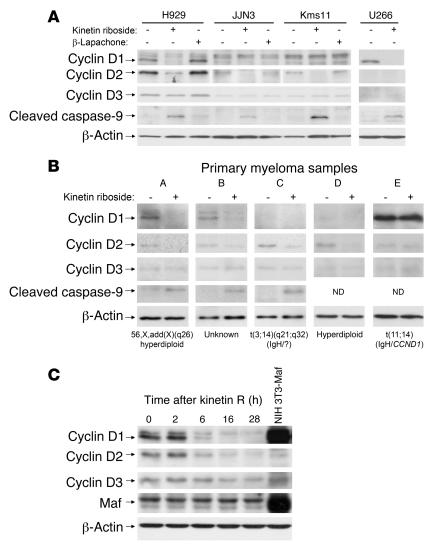
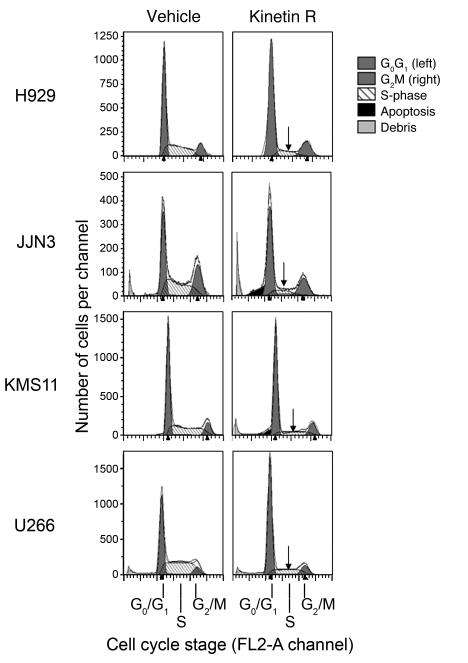
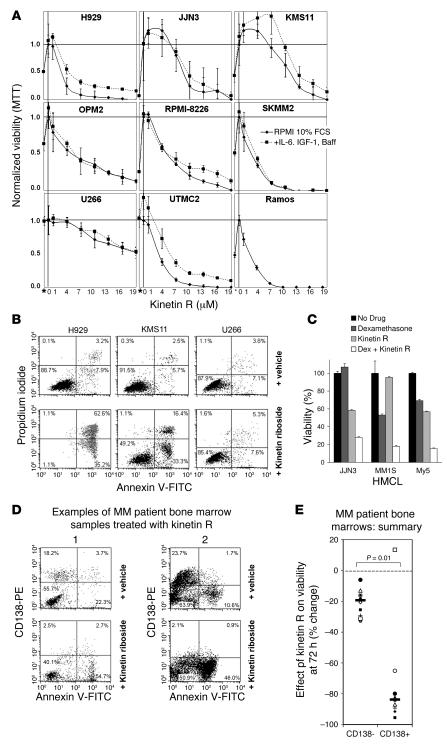
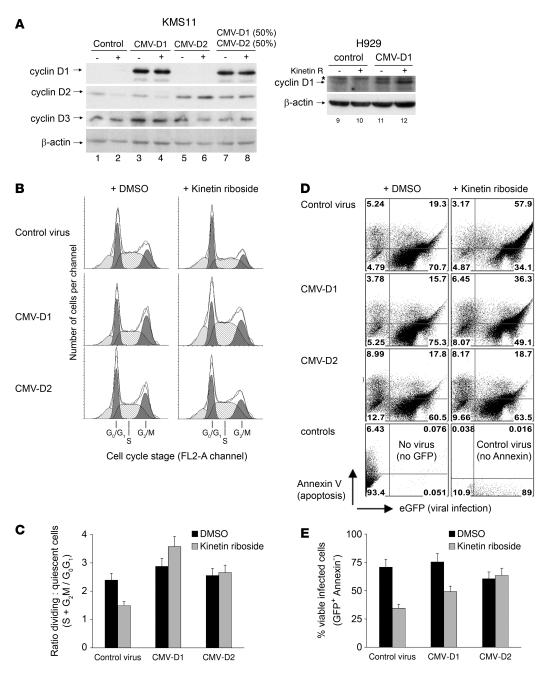
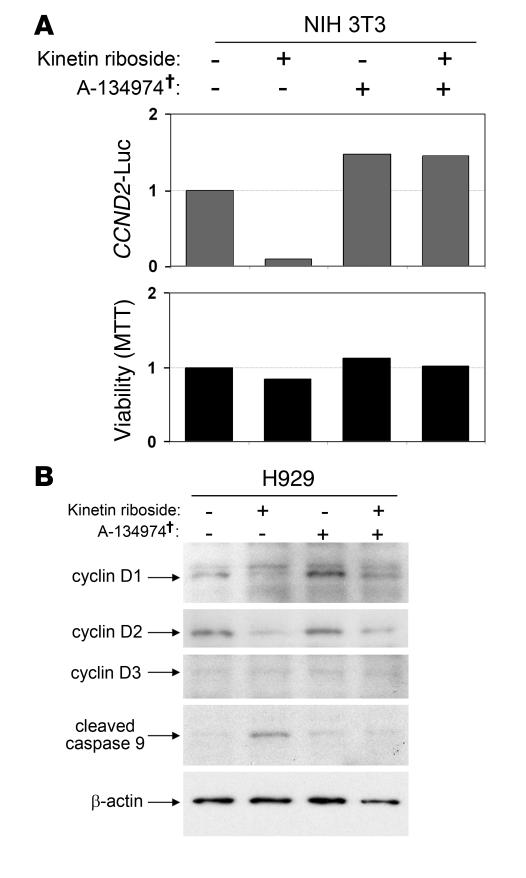
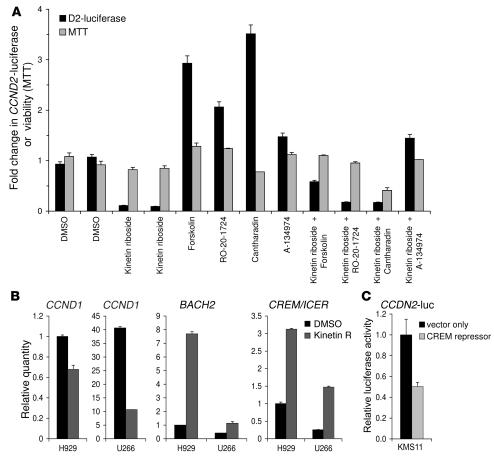
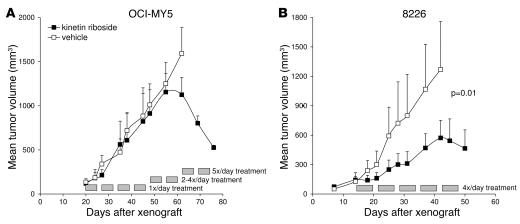
Knockout and transgenic studies in mice demonstrate that normal somatic tissues redundantly express 3 cyclin D proteins, whereas tumor cells seem dependent on a single overexpressed cyclin D. Thus, selective suppression of the individual cyclin D deregulated in a tumor represents a biologically valid approach to targeted cancer therapy. In multiple myeloma, overexpression of 1 of the cyclin D proteins is a ubiquitous feature, unifying at least 7 different initiating genetic events. We demonstrate here that RNAi of genes encoding cyclin D1 and cyclin D2 (CCND1 and CCND2, respectively) inhibits proliferation and is progressively cytotoxic in human myeloma cells. By screening a chemical library using a cell-based assay for inhibition of CCND2 trans-activation, we identified the plant cytokinin kinetin riboside as an inhibitor of CCND2 trans-activation. Kinetin riboside induced marked suppression of CCND2 transcription and rapidly suppressed cyclin D1 and D2 protein expression in primary myeloma cells and tumor lines, causing cell-cycle arrest, tumor cell-selective apoptosis, and inhibition of myeloma growth in xenografted mice. Mechanistically, kinetin riboside upregulated expression of transcription repressor isoforms of cAMP-response element modulator (CREM) and blocked both trans-activation of CCND2 by various myeloma oncogenes and cis-activation of translocated CCND1, suggesting induction of an overriding repressor activity that blocks multiple oncogenic pathways targeting cyclin D genes. These data support targeted repression of cyclin D genes as a therapeutic strategy for human malignancies.
Figures
Similar articles
-
Post-transcriptional Modifications Contribute to the Upregulation of Cyclin D2 in Multiple Myeloma.Clin Cancer Res. 2016 Jan 1;22(1):207-17. doi: 10.1158/1078-0432.CCR-14-2796. Epub 2015 Sep 4. Clin Cancer Res. 2016. PMID: 26341922
-
A small-molecule inhibitor of D-cyclin transactivation displays preclinical efficacy in myeloma and leukemia via phosphoinositide 3-kinase pathway.Blood. 2011 Feb 10;117(6):1986-97. doi: 10.1182/blood-2010-05-284810. Epub 2010 Dec 6. Blood. 2011. PMID: 21135258
-
Cyclin D type does not influence cell cycle response to DNA damage caused by ionizing radiation in multiple myeloma tumours.Br J Haematol. 2016 Jun;173(5):693-704. doi: 10.1111/bjh.13982. Epub 2016 May 4. Br J Haematol. 2016. PMID: 27146121
-
Critical roles for immunoglobulin translocations and cyclin D dysregulation in multiple myeloma.Immunol Rev. 2003 Aug;194:96-104. doi: 10.1034/j.1600-065x.2003.00052.x. Immunol Rev. 2003. PMID: 12846810 Review.
-
[Molecular mechanisms controlling the cell cycle: fundamental aspects and implications for oncology].Cancer Radiother. 2001 Apr;5(2):109-29. doi: 10.1016/s1278-3218(01)00087-7. Cancer Radiother. 2001. PMID: 11355576 Review. French.
Cited by
-
The natural pesticide dihydrorotenone induces human plasma cell apoptosis by triggering endoplasmic reticulum stress and activating p38 signaling pathway.PLoS One. 2013 Jul 26;8(7):e69911. doi: 10.1371/journal.pone.0069911. Print 2013. PLoS One. 2013. PMID: 23922854 Free PMC article.
-
Glucose Induces Mouse β-Cell Proliferation via IRS2, MTOR, and Cyclin D2 but Not the Insulin Receptor.Diabetes. 2016 Apr;65(4):981-95. doi: 10.2337/db15-0529. Epub 2016 Jan 6. Diabetes. 2016. PMID: 26740601 Free PMC article.
-
Measures of association for identifying microRNA-mRNA pairs of biological interest.PLoS One. 2012;7(1):e29612. doi: 10.1371/journal.pone.0029612. Epub 2012 Jan 11. PLoS One. 2012. PMID: 22253745 Free PMC article.
-
Harmol hydrochloride dihydrate induces autophagy in neuro cells and promotes the degradation of α-Syn by Atg5/Atg12-dependent pathway.Food Sci Nutr. 2022 Aug 12;10(12):4371-4379. doi: 10.1002/fsn3.3031. eCollection 2022 Dec. Food Sci Nutr. 2022. PMID: 36514773 Free PMC article.
-
Potential Therapeutics Targeting Upstream Regulators and Interactors of EHMT1/2.Cancers (Basel). 2022 Jun 9;14(12):2855. doi: 10.3390/cancers14122855. Cancers (Basel). 2022. PMID: 35740522 Free PMC article. Review.
References
-
- [No authors listed]. 2004. Cancer facts and figures 2004. American Cancer Society. Atlanta, Georgia, USA. 58 pp.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
