

Strict regulation of gene expression from a high-copy plasmid utilizing a dual vector system
- PMID: 18434195
- PMCID: PMC2442401
- DOI: 10.1016/j.pep.2008.03.014
Strict regulation of gene expression from a high-copy plasmid utilizing a dual vector system
Abstract
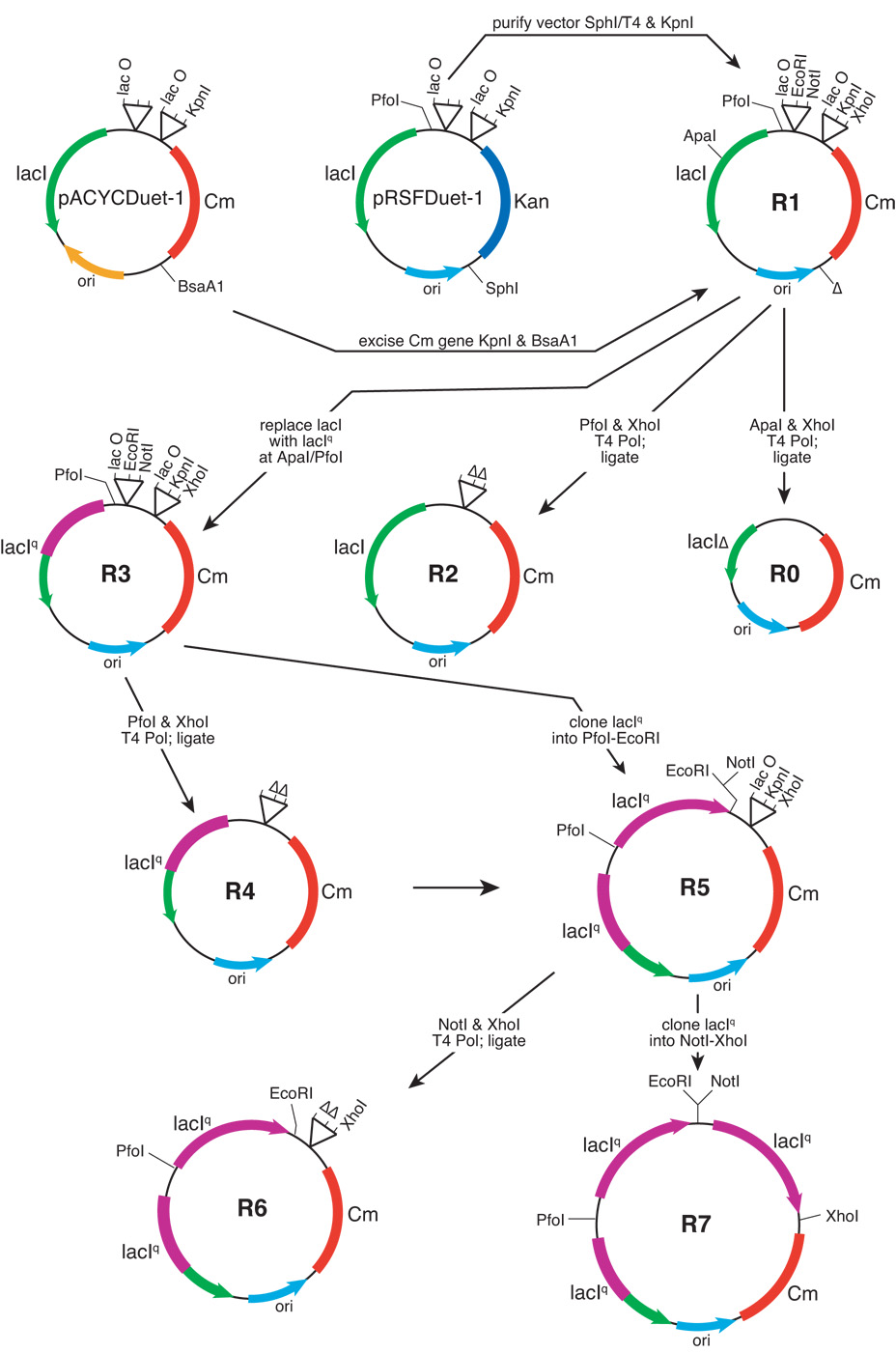
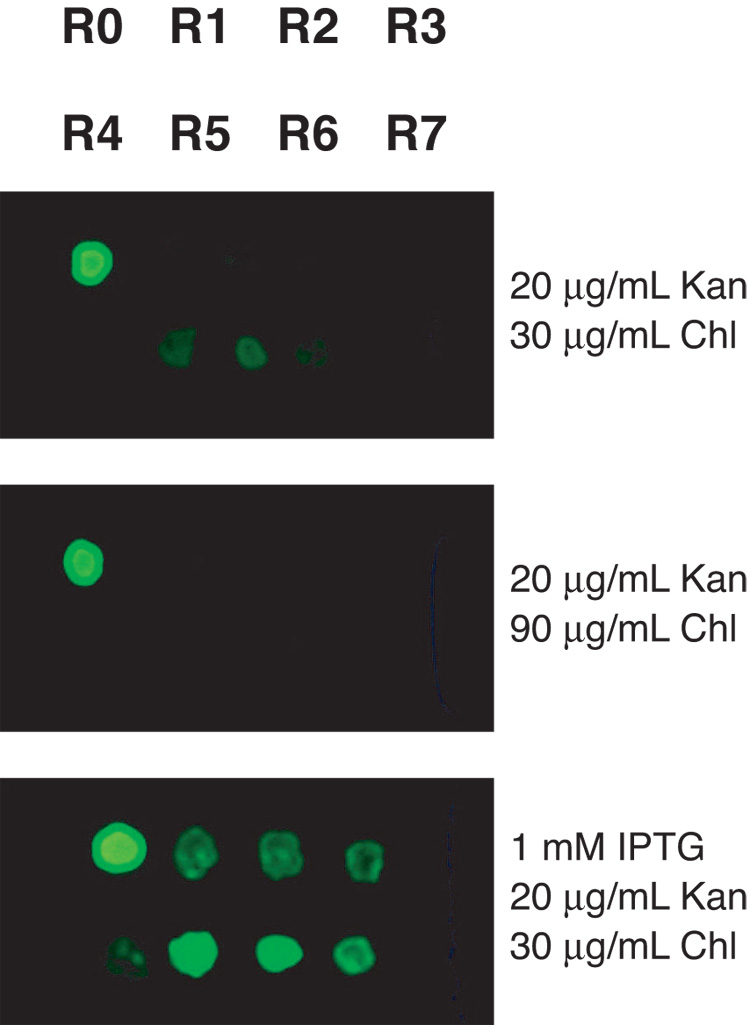
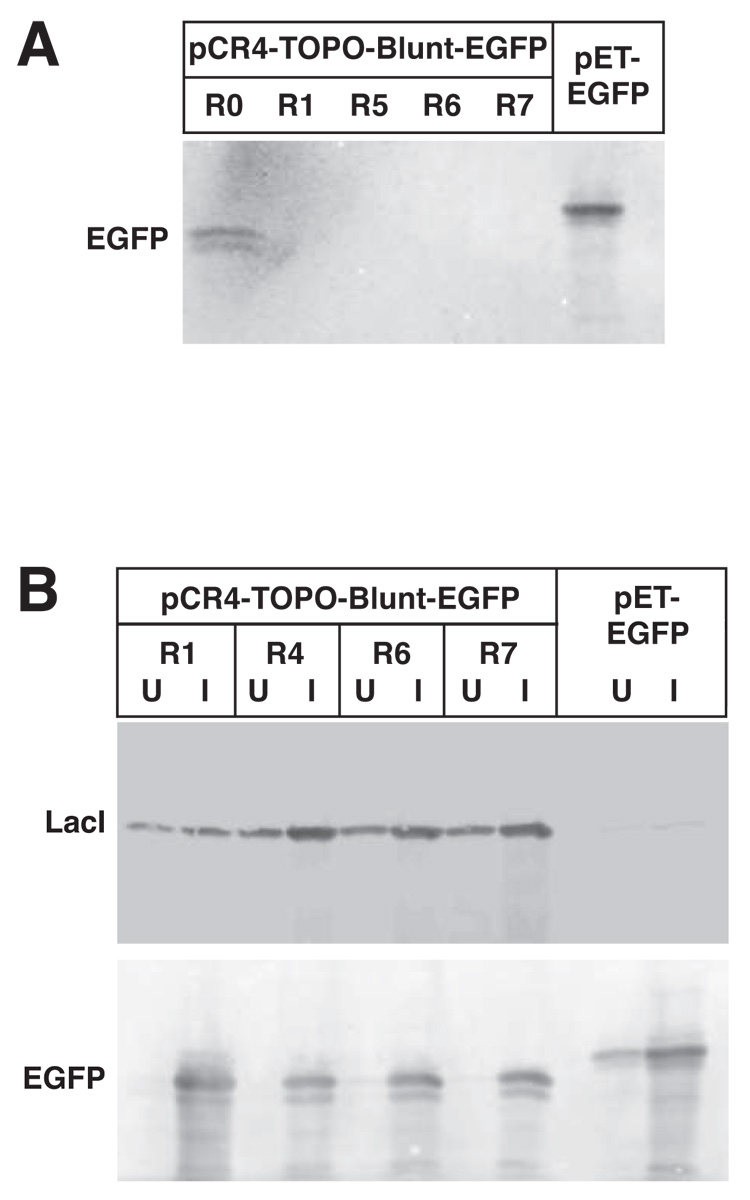
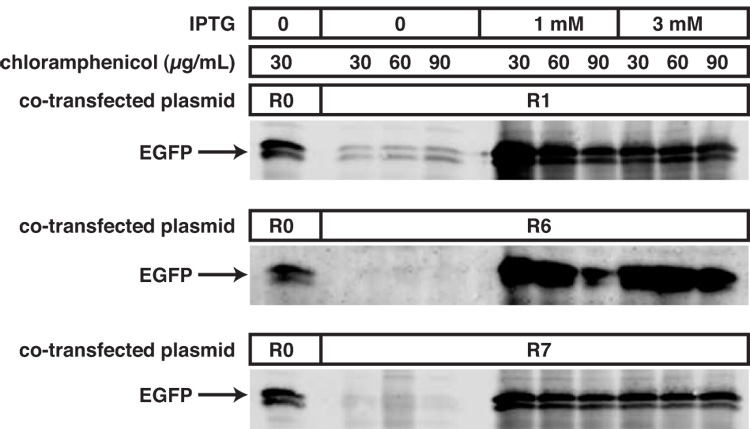
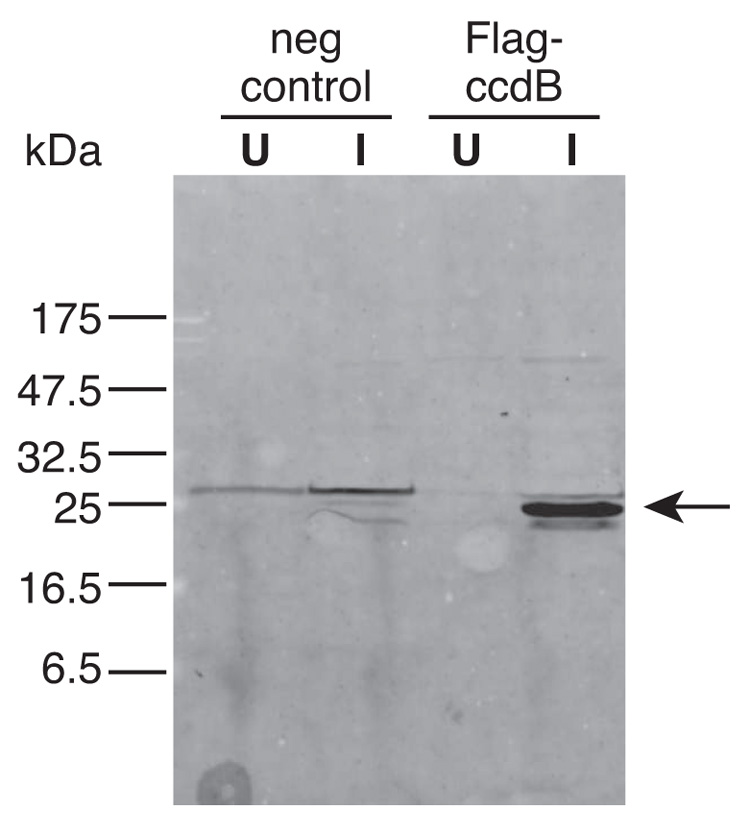
High-copy plasmids are useful for producing large quantities of plasmid DNA, but are generally inadequate for tightly regulating gene expression. Attempts to suppress expression of genes on high-copy plasmids often results in residual or "leaky" production of protein. For stringent regulation of gene expression, it is often necessary to excise the gene of interest and subclone it into a low-copy plasmid. Here, we report a dual plasmid technique that enables tight regulation of gene expression driven by the lac promoter in a high-copy vector. A series of plasmids with varying copies of the lacI(q) gene have been constructed to permit titration of the LacI protein. When a high-copy plasmid is transformed along with the appropriate lacI(q)-containing plasmid, tight gene regulation is achieved, thus eliminating the need to subclone genes into low-copy plasmids. In addition, we show that this dual plasmid technique enables high-copy gene expression of a protein lethal to Escherichia coli, the ccdB protein. In principle, this technique can be applied to any high-copy plasmid containing the popular pUC replication of origin and provides an easier means of obtaining rigid control over gene expression.
Figures
References
-
- Baneyx F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999;10:411–421. - PubMed
-
- Munson M, Predki PF, Regan L. ColE1-compatible vectors for high-level expression of cloned DNAs from the T7 promoter. Gene. 1994;144:59–62. - PubMed
-
- Warne SR, Thomas CM, Nugent ME, Tacon WC. Use of a modified Escherichia coli trpR gene to obtain tight regulation of high-copy-number expression vectors. Gene. 1986;46:103–112. - PubMed
-
- Lee CL, Ow DS, Oh SK. Quantitative real-time polymerase chain reaction for determination of plasmid copy number in bacteria. J. Microbiol. Methods. 2006;65:258–267. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
