

Species divergence and the measurement of microbial diversity
- PMID: 18435746
- PMCID: PMC2443784
- DOI: 10.1111/j.1574-6976.2008.00111.x
Species divergence and the measurement of microbial diversity
Abstract
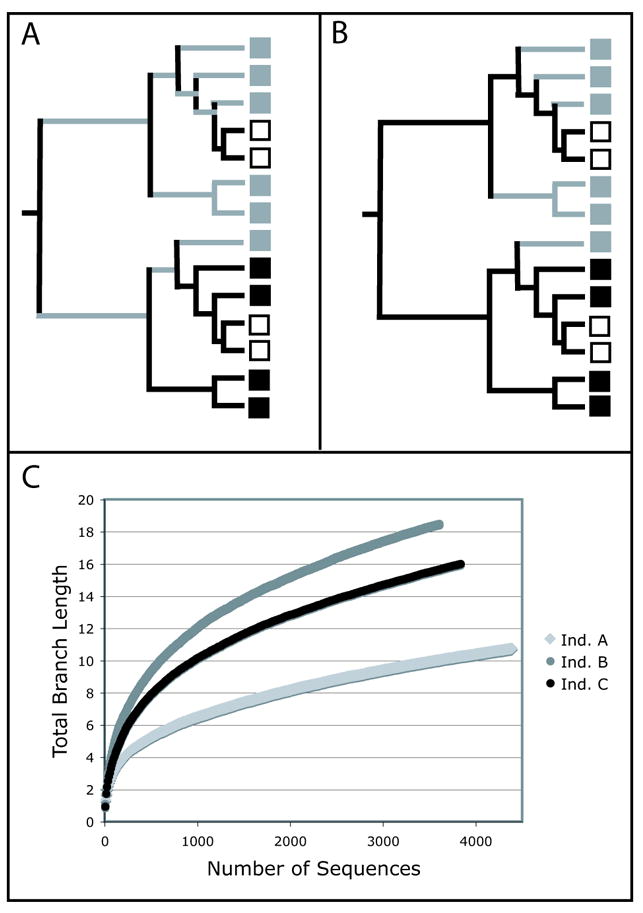
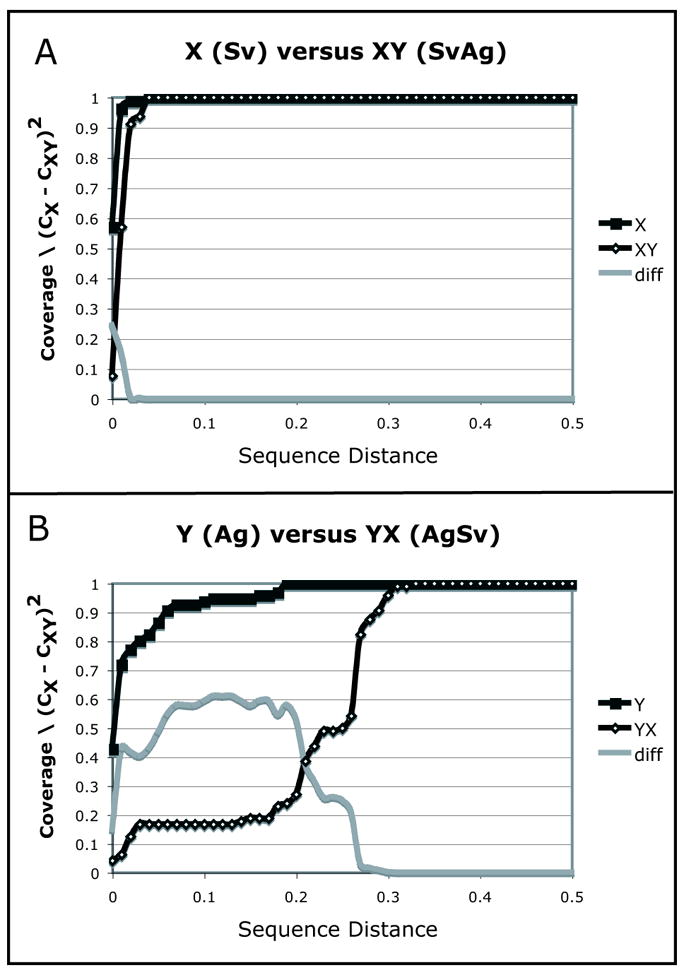
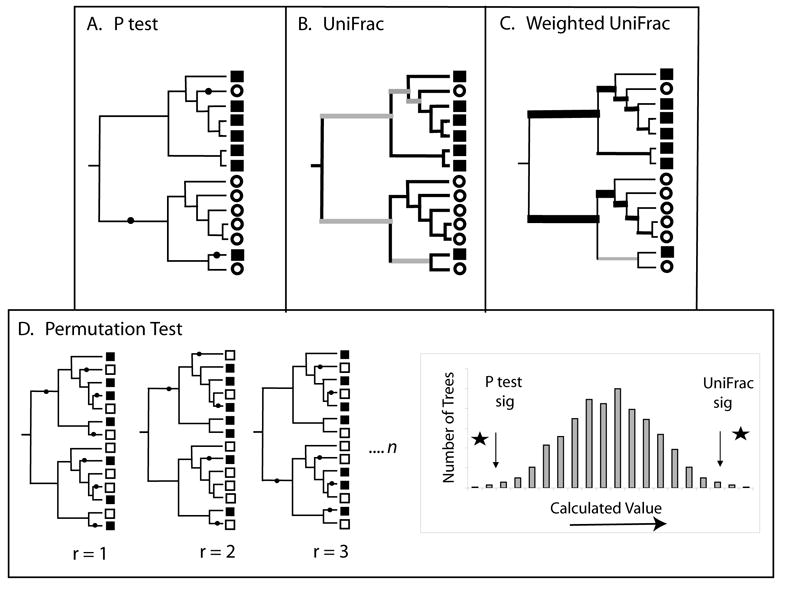
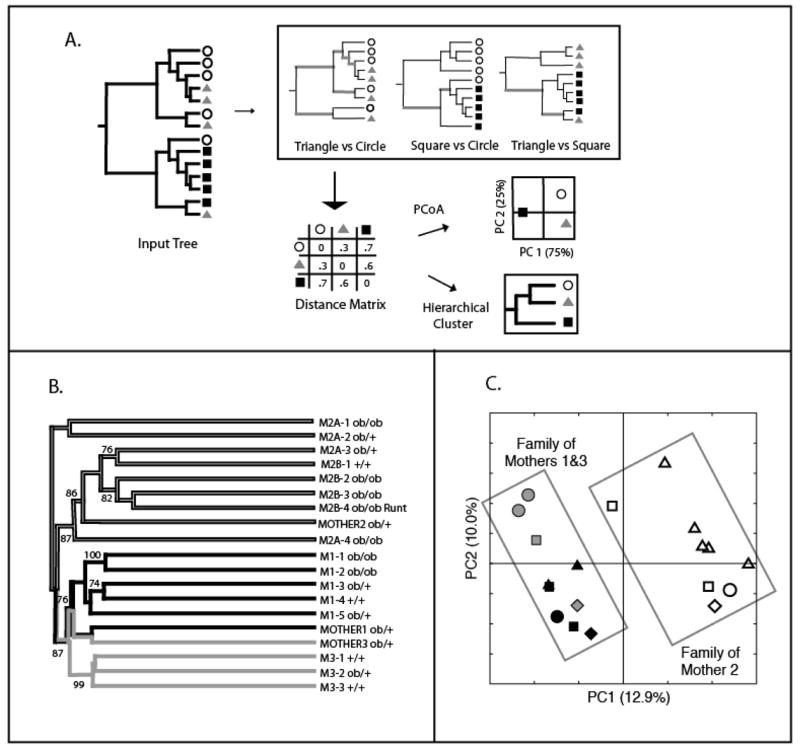
Diversity measurement is important for understanding community structure and dynamics, but has been particularly challenging for microorganisms. Microbial community characterization using small subunit rRNA (SSU rRNA) gene sequences has revealed an extensive, previously unsuspected diversity that we are only now beginning to understand, especially now that advanced sequencing technologies are producing datasets containing hundreds of thousands of sequences from hundreds of samples. Efforts to quantify microbial diversity often use taxon-based methods that ignore the fact that not all species are equally related, which can therefore obscure important patterns in the data. For example, alpha-diversity (diversity within communities) is often estimated as the number of species in a community (species richness), and beta-diversity (partitioning of diversity among communities) is often based on the number of shared species. Methods for measuring alpha- and beta-diversity that account for different levels of divergence between individuals have recently been more widely applied. These methods are more powerful than taxon-based methods because microorganisms in a community differ dramatically in sequence similarity, which also often correlates with phenotypic similarity in key features such as metabolic capabilities. Consequently, divergence-based methods are providing new insights into microbial community structure and function.
Figures
References
-
- Bass-Becking LGM. Geobiologie of Inleiding tot de Milieukunde. 1934. W.p van Stockum & Zoon N.V.
-
- Bohannan BJ, Hughes J. New approaches to analyzing microbial biodiversity data. Curr Opin Microbiol. 2003;6:282–287. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
