

Progress and challenges in protein structure prediction
- PMID: 18436442
- PMCID: PMC2680823
- DOI: 10.1016/j.sbi.2008.02.004
Progress and challenges in protein structure prediction
Abstract
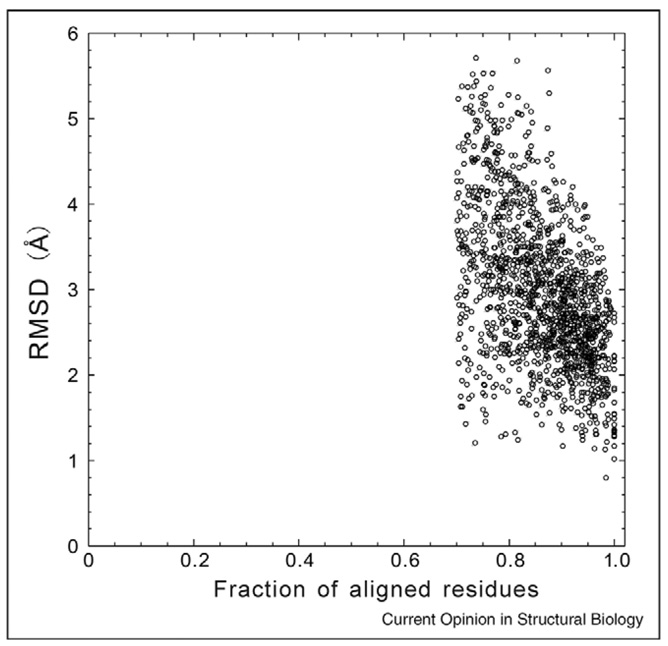
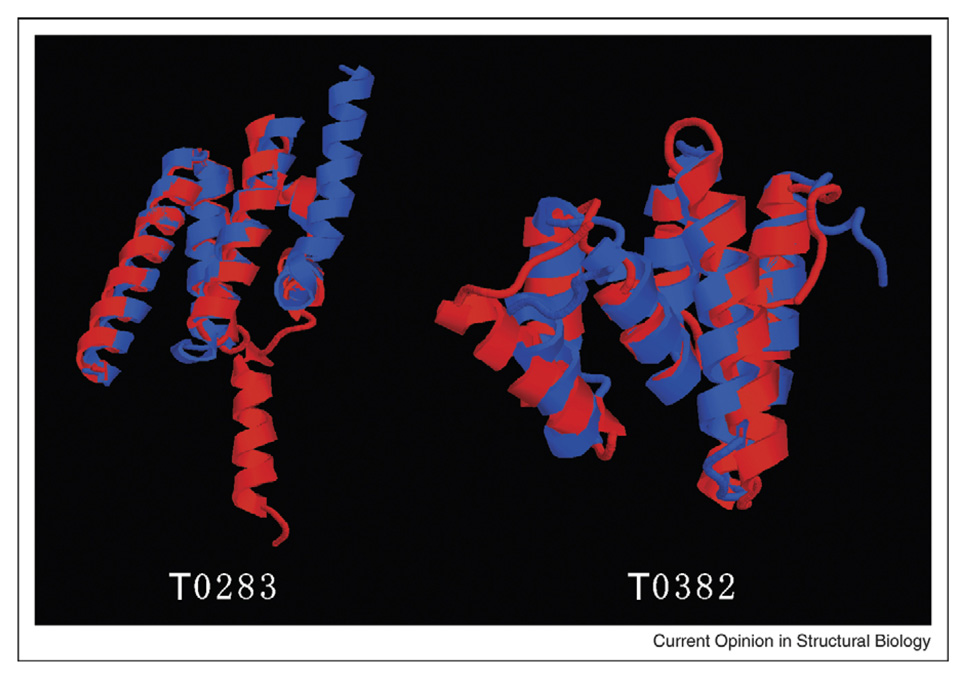
Depending on whether similar structures are found in the PDB library, the protein structure prediction can be categorized into template-based modeling and free modeling. Although threading is an efficient tool to detect the structural analogs, the advancements in methodology development have come to a steady state. Encouraging progress is observed in structure refinement which aims at drawing template structures closer to the native; this has been mainly driven by the use of multiple structure templates and the development of hybrid knowledge-based and physics-based force fields. For free modeling, exciting examples have been witnessed in folding small proteins to atomic resolutions. However, predicting structures for proteins larger than 150 residues still remains a challenge, with bottlenecks from both force field and conformational search.
Figures
References
-
- Burley SK, Almo SC, Bonanno JB, Capel M, Chance MR, Gaasterland T, Lin D, Sali A, Studier FW, Swaminathan S. Structural genomics: beyond the human genome project. Nat Genet. 1999;23:151–157. - PubMed
-
-
Chandonia JM, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311:347–351. The authors review and assess the gain and loss of the structural genomics project in the past five years in contrast with traditional structural biology.
-
-
-
Pieper U, Eswar N, Davis FP, Braberg H, Madhusudhan MS, Rossi A, Marti-Renom M, Karchin R, Webb BM, Eramian D, et al. MODBASE: a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2006;34:D291–D295. MODBASE is a database of 3D models built by the MODELLER pipeline for all protein sequences in SwissProt based on available structural templates in the PDB library.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
